一个常染色体显性遗传性聋家系致病基因鉴定△
2021-01-15曹婧媛袁阳程静卢宇杨长亮阳光杨慧周佳吴雄英袁慧军孙艺
曹婧媛 袁阳 程静 卢宇 杨长亮 阳光 杨慧 周佳 吴雄英 袁慧军 孙艺
耳聋是人类最常见的感觉缺陷性疾病,可导致严重的言语交流障碍,由遗传因素和环境因素(如噪声、创伤、药物等) 共同作用所致[1]。常染色体显性遗传性聋新基因的传统鉴定方式主要是利用大家系进行连锁分析和候选基因的全序列测序;由于需要家系中有较多的耳聋患者,以及检测和分析方法效率较低,新基因的鉴定往往需要数年的研究。近十年来随着高通量测序技术的进步,遗传性聋的致病基因研究飞速发展,越来越多的常染色体显性遗传性聋新基因利用高通量测序技术被鉴定,如:REST[2]、NLRP3[3]、COL11A[4]、PTPRQ[5]、DMXL2[6]等,该技术因其简便、经济、准确、高效的优势成为遗传性聋基因研究的最主流方法。本研究对一来自河南省的遗传性聋家系进行深入的临床调查和基因研究,并运用高通量测序和传统的连锁分析技术研究其致病基因,报告如下。
1 资料与方法
1.1家系资料的采集及临床听力学检查 本研究收集到一个遗传性聋家系(编号HBSY-012),家系先证者就诊于中国人民解放军中部战区总医院耳鼻喉科门诊,完成听力学检测及全身体格检查,包括纯音听阈检测、声导抗、耳声发射、听性脑干反应及颞骨CT等,明确是否存在中耳、内耳畸形、颅内占位病变以及全身其它系统的异常。
经医院医学伦理委员会同意后,由耳鼻喉科的家系调查小组人员前往家系所在地进行调查,所有参加本项目的家系成员均签署知情同意书(未成年人由监护人代签),然后进行详细的病史询问、填写问诊表,全身体格检查,进行纯音听阈和声导抗测试,并抽取10 ml外周血液样本。
1.2诊断标准 耳聋表型的判断标准参照《关于非综合征型遗传性聋家系遗传学及听力学描述术语建议案》[2]。
1.3家系样本DNA的提取 该家系现存三代34人,共采集到28人血液样本;对采集到的28例家系成员(18例正常听力者,10例耳聋者)的外周血进行基因组DNA提取(Axygen公司AxyPrep血基因组DNA中量试剂盒)。
1.4高通量测序 采用Illumina公司的HiSeq2000测序平台进行高通量测序(北京迈基诺公司),首先对先证者进行162个已知遗传性聋相关基因靶向测序(基因信息见表1),随后完成全外显子组测序 (Aligent Exome v5捕获试剂盒),平均深度超过200X。
1.4.1测序数据分析 全外显子组测序后得到的数据进行生物信息学分析,测序质控标准:Q20平均比例在90%以上、Q30平均比例在85%以上以及GC 含量分布无明显偏移。测序数据分析获得的3个样本的变异根据样本表型进行共分离过滤,通过gnomAD(MAF>0.1%)过滤高频变异位点,通过MGI数据库、JacksonLab数据库等耳聋模式动物和功能研究数据库,筛选与听觉功能相关的候选基因,通过PolyPhen2、SIFT等软件预测候选变异的致病性。
1.4.2Sanger测序进行家系验证 全外显子组测序结果经过生物信息学分析得到可疑致病位点,对家系中表型明确的家系成员的基因组DNA进行PCR扩增,扩增产物纯化后进行Sanger测序验证,明确该可疑位点是否在家系中共分离。
1.5连锁分析 对参与连锁分析的20例家系成员的每名参与者提取20 ng基因组DNA,采用Illumina Infinium Human OminiZhongHua-8全基因组芯片进行实验(上海伯豪生物技术有限公司),通过优势对数记分法的LOD(likelihood of odds)值评判连锁的概率。
1.5.1数据分析 在进行连锁分析前,首先对原始数据位点进行质控,包括去除等位基因频率过低的、检出率过低的、不符合孟德尔遗传规律的位点,并将单核苷酸多态(single nucleotide polymorphism,SNP)作为标记进行连锁分析,每0.3 cM挑选一个位点。
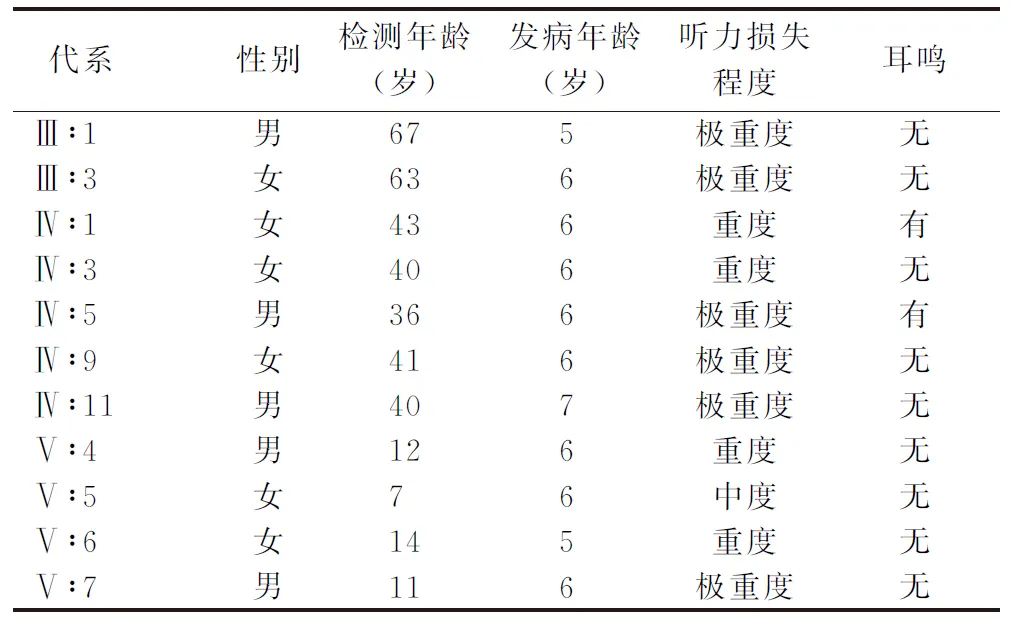
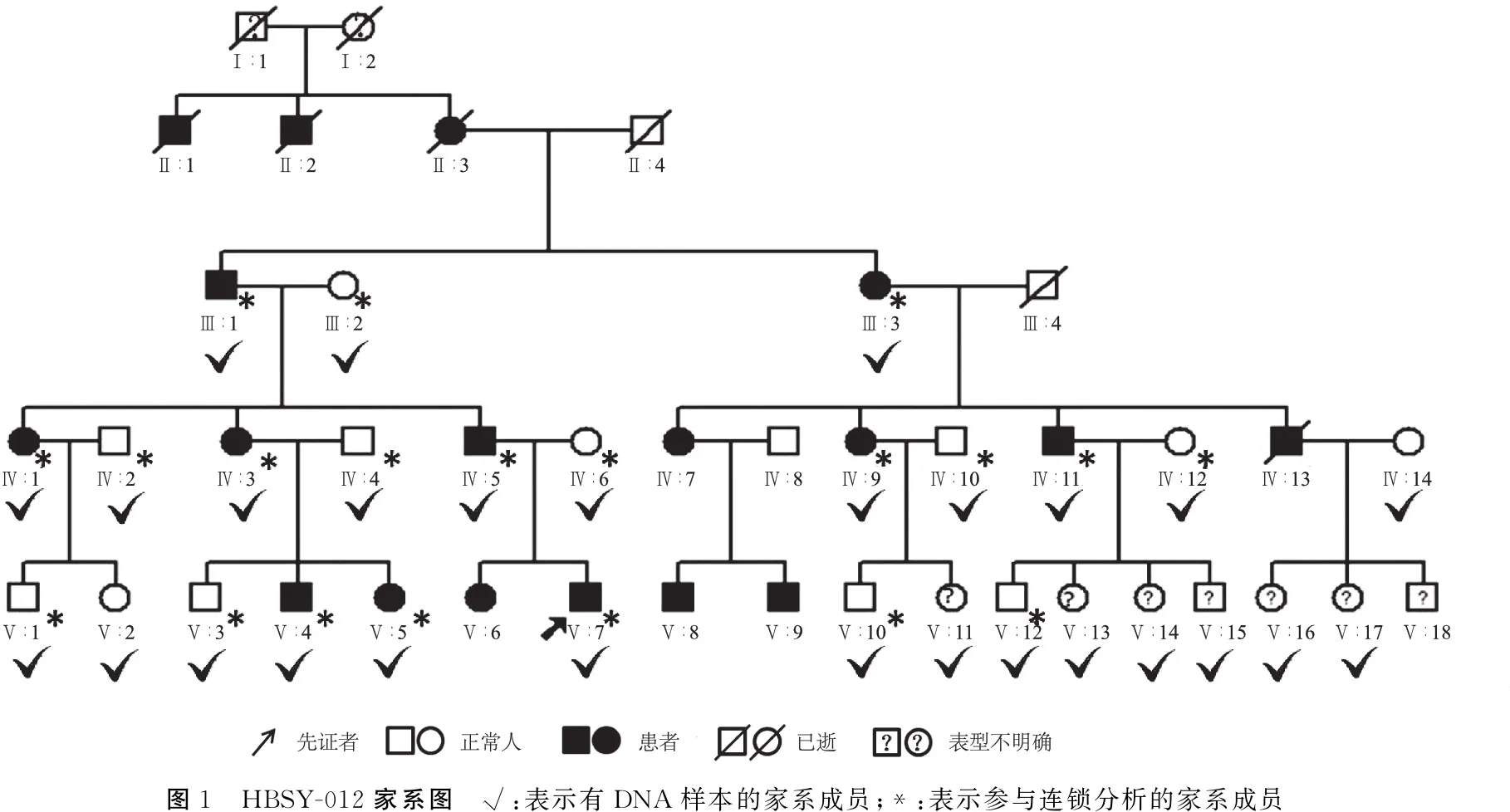
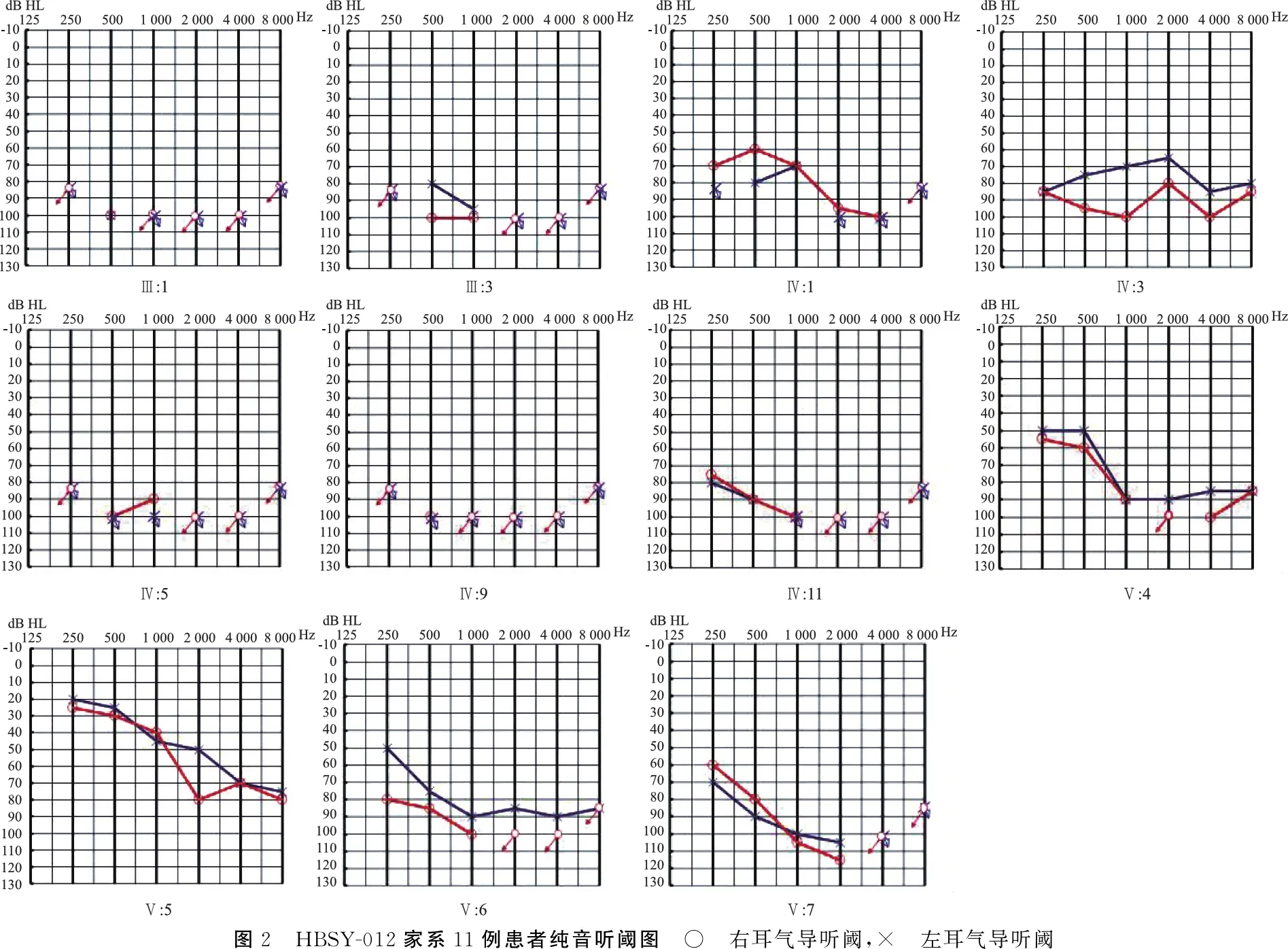
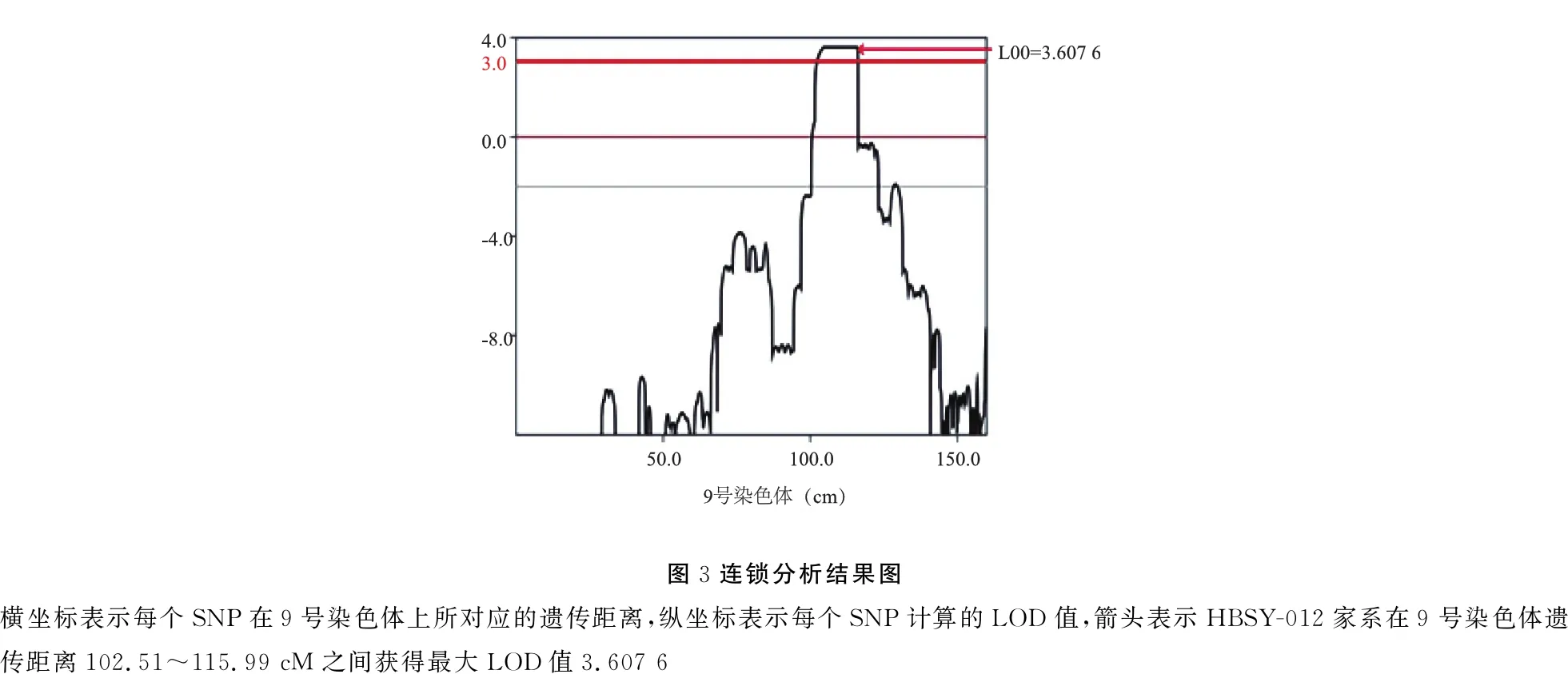
1.5.2家系连锁分析 使用软件merlin(v1.1.2)进行连锁分析,采用多点参数连锁分析模型。通常,LOD值≥3认为确定连锁,LOD值≥2则可能连锁,-2 表1 靶向测序的耳聋相关基因分类、名称及定位 2.1该家系的临床表型特征 HBSY-012家系来自河南省某县,现存三代共34人,14人诊断为感音神经性聋,除3人(Ⅳ∶7、Ⅴ∶8及Ⅴ∶9)未接受检测之外,其余11人均接受了详细听力学检查。该家系的14例患者均在5~7岁开始出现听力下降,为语后聋,患者中年龄最大66岁,最小7岁;9例患者仅有耳聋症状,2例患者伴耳鸣,无眩晕等前庭功能障碍,无耳毒性药物及环境噪声接触史,否认外伤史,其他系统均无异常。家系中感音神经性聋患者临床资料见表2。 表2 家系中感音神经聋患者性别、检测及发病年龄、听力损失程度及伴耳鸣情况 2.2该家系遗传学及耳聋表型特征 2.2.1家系遗传学特征分析 HBSY-012家系图(图1)遗传学特点:①四代连续发病;②每一代中男女均可患病;③男女患者均能将疾病传给下一代;④每一例耳聋患者的双亲中均有一例耳聋患者;⑤家系发病率高,第二代至第四代耳聋患病率为100%;⑥第五代中Ⅴ∶11、Ⅴ∶13、Ⅴ∶14、Ⅴ∶15、Ⅴ∶16、Ⅴ∶17、Ⅴ∶18未达到发病年龄(5~7岁),暂未出现听力下降情况,将其表型归为不确定。该家系符合典型常染色显性遗传特点。 2.2.2HBSY-012家系的耳聋表型特征 以患者Ⅴ∶5为例,患儿7岁,6岁时出现听力下降,表现为以高频下降为主的中度感音神经性聋,双耳对称,因其为家系患者中年龄最小者,代表该家系最初的发病特点。先证者Ⅴ∶7,年龄11岁,6岁时出现听力下降,现已发展为全频受累的双耳极重度感音神经性聋。家系第Ⅲ、Ⅳ代成员中耳聋者均表现为全频下降的重度或极重度感音神经性聋。该家系的耳聋表型特点是迟发性语后聋,耳聋的进展速度快,程度重。家系中耳聋患者纯音听阈图见图2。 2.3连锁分析结果 通过对20例家系成员(图1中*表示参与连锁分析的家系成员)的全基因组扫描SNP位点分型,使用软件merlin(v1.1.2)进行连锁分析,得到11 152个有效tagSNPs数量,平均遗传距离0.3 cM,LOD值>3的位点分布在9号染色体长臂q31.1-q31.3上,遗传标记SNP-A-rs4273907至SNP-A-rs10981098之间,遗传距离102.51~115.99 cM(物理位置102 977 295~114 591 623),其中最大LOD值3.607 6,支持肯定连锁(图3)。而其它染色体上均未发现较强的连锁关系。 图1 HBSY-012家系图 √:表示有DNA样本的家系成员;*:表示参与连锁分析的家系成员 图2 HBSY-012家系11例患者纯音听阈图 ○ 右耳气导听阈,× 左耳气导听阈 2.4高通量测序结果 首先,选取先证者Ⅴ∶7进行162个已知耳聋基因筛查,未发现致病突变。然后,选取家系中Ⅳ∶9、Ⅴ∶3、Ⅴ∶4进行全外显子组测序,其中Ⅳ∶9及Ⅴ∶4为家系中的耳聋患者,Ⅴ∶3为家系中的正常人。筛选Ⅳ∶9及Ⅴ∶4共有的杂合变异,过滤Ⅴ∶3携带的变异,共获得2 924个候选变异,通过gnomAD过滤高频变异后,在连锁分析定位的第9号染色体q31.1-q31.3区间内未发现候选变异,在该区间以外的候选变异中,通过MGI数据库、JacksonLab数据库等耳聋模式动物和功能研究数据库,筛选出可能与听觉功能相关的4个候选基因,候选变异为ANKMY2基因NM_020319:c.822_826del、DDX49基因NM_019070:c.341C>T、DEFB129基因NM_080831:c.284G>T以及EVI5基因NM_005665:c.2399C>T,共4个候选基因变异位点(图4)。 图3连锁分析结果图横坐标表示每个SNP在9号染色体上所对应的遗传距离,纵坐标表示每个SNP计算的LOD 值,箭头表示HBSY-012家系在9 号染色体遗传距离102.51~115.99 cM之间获得最大LOD值3.607 6 图4 Sanger测序图a.ANKMY2基因c.822_826del变异; b. DDX49基因c.341C>T变异; c.DEFB129基因c.284G>T变异; d.EVI5基因c.2399C>T变异 将上述4个候选基因变异位点分别进行家系验证,结果4个候选基因变异位点在家系中均无共分离现象,提示都不是家系的致病突变。 3.1HBSY-012家系临床表型特征分析 HBSY-012家系的发病年龄5~7岁,早期以高频下降为主,听力损失的特点是进展速度快,程度重。家系中年龄最小的患者Ⅴ∶5, 年龄7岁,6岁时出现听力下降,表现为以高频下降为主的中度感音神经性耳聋;先证者Ⅴ∶7,年仅11岁,6岁时出现听力下降,现已达极重度听力损失,且第Ⅲ、Ⅳ代患者均为全频下降的重度或极重度感音神经性聋。家系中参与研究的11例耳聋患者除了两例伴有耳鸣症状,其他患者耳聋为唯一症状,全身其他系统均正常,无耳毒性药物及环境噪声接触史。通过遗传图谱可发现,该家系共5代,现存3代共计34人,耳聋患者14人,第Ⅱ代至第Ⅳ代直系血缘关系亲属耳聋患病率为100%,男女均有患病,符合典型的常染色体显性遗传性非综合征型聋。 3.2致病基因的鉴定 耳聋基因鉴定的主要方式有家系连锁分析法和高通量测序法。家系连锁分析法在高通量测序出现之前曾被作为最主流的方法,已被证实是寻找家系耳聋致病基因最准确的方法之一,适用于表型明确的耳聋大家系,是从表型到基因型的正向研究。通过家系成员连锁分析,可将致病基因确定在染色体上的某个区间内,再逐步对区间内的基因进行排查;但连锁区域内可能多达上百个基因需要逐个进行突变检测,工作量庞大为其缺点。高通量测序技术的发展有效地解决了上述技术难点,2011年,Zheng等[8]将其课题组于1995年连锁分析定位的DFNA4(19q12-13.4)家系进行全外显子组测序,确定了该家系的致病基因CEACAM16。 目前遗传性聋基因研究应用的高通量测序主要包括耳聋相关基因靶向测序、全外显子组测序及全基因组测序。耳聋相关基因靶向测序是将目前已知的耳聋相关基因集中在一个panel中进行检测,成本低,耗时短,生物信息学分析较简便。全外显子组测序采用序列捕获技术将基因组DNA全部外显子区域一次性捕获下来,然后进行集中测序。外显子区为蛋白编码区,虽然只占到人类基因组的1%,但其中包含基因组DNA上所有蛋白质的编码序列[9]。研究表明,遗传性聋绝大多数为单基因病,符合孟德尔遗传规律,大部分孟德尔遗传病的致病突变均位于外显子区域[10]。目前认为全外显子组测序为鉴定遗传性聋致病基因最有效的方法,近年来就有多个直接利用全外显子组测序技术鉴定的耳聋新基因,如:LMX1A[11]、IFNLR1[12]、MYO3A[13]、PDE1C[14]、ESRP1[15]等。全基因组测序将外显子区域及内含子区域同时捕获,可以对外显子组区域以外的区域进行有效的基因检测,对于部分不能通过全外显子组测序鉴定出致病基因的家系或散发病例还需要全基因组测序进行更全面的基因检测。 本研究首先选取先证者Ⅴ∶7进行162个已知遗传性聋相关基因靶向测序,结果未检测出致病突变。分析可能的原因为致病基因未包括在所检测的基因中或为尚未发现的耳聋基因,抑或突变位点位于非编码区域。然后,本研究采取高通量测序和连锁分析相结合的方法探索HBSY-012家系的耳聋致病基因,对20名家系成员进行全基因组扫描SNP位点分型,结果进行连锁分析,LOD值>3的位点分布在9号染色体q31.1-q31.3上,遗传标记SNP-A-rs4273907至SNP-A-rs10981098之间,遗传距离为102.51~115.99 cM (物理位置为102 977 295~114 591 623),其中最大LOD值3.607 6;将该家系的致病基因定位于第9号染色体q31.1-q31.3区间,与已知耳聋基因座位DFNA56(9q31.3-q34.3)毗邻;2013年Zhao等[16]已将DFNA56家系应用全外显子组测序,明确了家系致病基因TNC。 本研究选取家系中Ⅳ∶9、Ⅴ∶3、Ⅴ∶4进行全外显子组测序,Ⅳ∶9及Ⅴ∶4为家系中的耳聋患者,Ⅴ∶3作为家系中的正常人,共获得2 924个候选变异,通过gnomAD过滤高频变异后,在连锁分析定位的第9号染色体q31.1-q31.3区间内未发现致病突变;在该区间以外的候选变异中,通过MGI数据库、JacksonLab数据库等耳聋模式动物和功能研究数据库,筛选出可能与听觉功能相关的4个候选基因,候选变异为ANKMY2基因NM_020319:c.822_826del、DDX49基因NM_019070:c.341C>T、DEFB129基因NM_080831:c.284G>T以及EVI5基因NM_005665:c.2399C>T共4个候选基因变异位点;将上述4个候选基因变异位点分别进行家系验证,结果该4个候选基因变异位点在家系中均无共分离现象,提示都不是家系的致病突变。 因此,考虑该家系的致病突变可能来源于基因的非编码区域或者罕见的CNV/SV,拟进一步扩大寻找范围进行全基因组测序,以鉴定致病基因突变。
2 结果

3 讨论
