Characterization and comparison of chloroplast genomes from two sympatric Hippophae species (Elaeagnaceae)
2021-01-11LuoyunWangJingWangCaiyunHeJianguoZhangYanfeiZeng
Luoyun Wang · Jing Wang · Caiyun He ·Jianguo Zhang · Yanfei Zeng
Abstract The genus Hippophae includes deciduous shrubs or small trees, which provide many ecological, economic,and social benef its. We assembled and annotated the chloroplast genomes of sympatric Hippophae gyantsensis (Rousi)Lian and Hippophae rhamnoides Linn subsp. yunnanensis Rousi and comparatively analyzed their sequences. The fulllength chloroplast genomes of H. gyantsensis and H. rhamnoides subsp. yunnanensis were 155,260 and 156,415 bp,respectively; both featured a quadripartite structure with two copies of a large inverted repeat (IR) separated by small(SSC) and large (LSC) single-copy regions. Each Hippophae chloroplast genome contained 131 genes, comprising 85 protein-coding, 8 ribosomal RNA, and 38 transfer RNA genes.Of 1302 nucleotide substitutions found between these two genomes, 824 (63.29%) occurred in the intergenic region or intron sequences, and 478 (36.71%) were located in the coding sequences. The SSC region had the highest mutation rate, followed by the LSC region and IR regions. Among the protein-coding genes, three had a ratio of nonsynonymous to synonymous substitutions (Ka/Ks) > 1 yet none were significant, and 66 had Ka/Ks < 1, of which 46 were significant. We found 20 and 16 optimal codons, most of which ended with A or U, for chloroplast protein-coding genes of H. gyantsensis and H. rhamnoides subsp. yunnanensis , respectively.Phylogenetic analysis of five available whole chloroplast genome sequences in the family Elaeagnaceae-using one Ziziphus jujube sequence as the outgroup-revealed that all five plant species formed a monophyletic clade with two subclades: one subclade consisted of three Hippophae species, while the other was formed by two Elaeagnus species,supported by 100% bootstrap values. Together, these results suggest the chloroplast genomes among Hippophae species are conserved, both in structure and gene composition, due to general purifying selection; like many other plants, a significant AT preference was discerned for most proteincoding genes in the Hippophae chloroplast genome. This study provides a valuable reference tool for future research on the general characteristics and evolution of chloroplast genomes in the genus Hippophae.
Keywords Chloroplast genome · Hippophae gyantsensis(Rousi) Lian · Hippophae rhamnoides Linn subsp.yunnanensis · Ka/Ks · Optimal codon
Introduction
Green plants and algae rely on their chloroplasts for energy conversion and photosynthesis (Ruhlman and Jansen 2014;Nazareno et al. 2015). These semi-autonomous cellular organelles house relatively independent genetic material, called chloroplast DNA (Sugiura 1992), inherited in a maternal fashion in approximately 80% of angiosperms (Ruhlman and Jansen 2014). The chloroplast genome of most plants occurs in high copy numbers in each cell, with relatively little variation in gene content and order (Ruhfel et al.2014). Most chloroplast genomes of plants have a cyclic structure that includes one large single-copy (LSC) region,two inverted repeat (IR) regions, and one small single-copy(SSC) region, whose full length amounts to 120-2500 kb,with 110-130 genes (Zhengqiu et al. 2006; Guisinger et al.2010; Wicke et al. 2011; Wang et al. 2012; Liu et al. 2018).With advances in molecular biology, especially the development oflarge-scale high-throughput genetic sequencing techniques, research on whole chloroplast genomes is increasingly intensive and widespread (Ding et al. 2017).
The genus Hippophae (Elaeagnaceae) includes seven recognized species and 11 subspecies (Bartish 2002). These plants usually grow as dioecious deciduous shrubs or small trees, that are widely distributed on the Eurasian continent (He et al. 2016) and highly adaptable to stress factors(Kortesniemi et al. 2017). For example, they can withstand extreme temperatures of − 43 to 40 °C, growing well in dry,sandy, rocky, saline, and ravine soils (Stobdan et al. 2008;Jadhav 2014). The Hippophae root system can form root nodules that fix nitrogen and thus improve soil fertility, a benef it for land reclamation (Zhou et al. 2017). Both the fruits and leaves of Hippophae are rich in bioactive substances such as carotene, various vitamins (A, B1, B2, P, C,K, and E), catechins, elagic acid, ferulic acid, and folic acid and in calcium, magnesium, and potassium-which may be benef icial to human health, indicating the vast potential of these plants as food and nutrition resources (Bal et al. 2011;Dhyani et al. 2011; Suryakumar and Gupta 2011; Qian and Jin 2015). Much research has analyzed the interspecific differentiation and genetic diversity of Hippophae using traditional methods, such as amplified fragment length polymorphism (AFLP) (Li and Yu 2008), simple sequence repeats(SSRs) (Li et al. 2017), internal transcribed spacers (ITSs),and chloroplast trnL- F and trnS- G sequences (Jia et al. 2012;Ma et al. 2014). However, few studies have investigated Hippophae at its chloroplast genome level (Chen and Zhang 2017), with no published work yet comparing the chloroplast genomes of sympatric species in this genus.
Two species in particular, Hippophae gyantsensis (Rousi)Lian and Hippophae rhamnoides Linn subsp. yunnanensis Rousi, occur on the Qinghai Tibetan Plateau, a high-elevation region highly sensitive to climate change (Cheng 2008).One study suggested alpine environments can change the chloroplast microstructure in plants (e.g., Lütz and Engel 2007), so an in-depth examination of chloroplast genomes from species on this plateau area is of great importance for understanding chloroplast evolution in sympatric species.
In this study, we completed random interrupt sequencing of the genomes for H. gyantsensis and H. rhamnoides subsp. yunnanensis via high-throughput sequencing, then de novo assembled, annotated and systematically compared the complete chloroplast genomes of the two Hippophae plants.We also revealed key chloroplast genome characteristics of Hippophae, which will inform future studies of chloroplast genomes in this genus and other angiosperms in the Qinghai Tibetan Plateau region.
Materials and methods
DNA extraction and sequencing
Leaves of two Hippophae species (H. gyantsensis and H.rhamnoides subsp. yunnanensis) were respectively collected from a single wild specimen growing in Gongbo’gyamda County, Linzhi City of Tibet Autonomous Region (3600 m a.s.l.; 93° 15′ E; 29° 48′ N; temperate semi-humid plateau monsoon climate) in August 2017, immediately dried with silica gel desiccant, and then taken to the laboratory. For each species, total DNA was extracted from 30 mg of dry leaves using the cetyltrimethylammonium bromide (CTAB)method (Doyle 1987). Agarose gel electrophoresis was used to assess the validity of this DNA extraction process. Total DNA was then sent to the Novogene Company (Beijing,China) for complete random interrupt sequencing using Illumina HiSeq and 150-bp paired-end sequencing libraries with an insert size of 350 bp. Finally, for each Hippophae species, a 35-G sequence data set for the nuclear genome was obtained with a depth of 30 ×.
Assembling complete chloroplast genomes
To assemble the complete chloroplast genomes, NOVOPlasty (v.2.7.0) software was used (Dierckxsens et al. 2017);it uses the raw data generated by Illumina platforms as an insert file and a short chloroplast DNA sequence as the seed file. The H. gyantsensis trnL- F sequence (KU304417) and the H. rhamnoides subsp. yunnanensis trnL- F sequence(KU304405), both downloaded from the National Center for Biotechnology Information (NCBI) website, were set as the seed sequences, respectively. NOVOPlasty parameters were as follows: K-mer = 39, type = ‘chloro’, genome range = 120,000-200,000; all other parameters were set to default values.
For estimating the assembly accuracy of the H. rhamnoides subsp. yunnanensis and H. gyantsensis chloroplast genomes, six chloroplast fragments showing large differences between the two assembled genomes were amplified and then sequenced using Sanger technology. PCR amplifications used primers designed from the currently assembled chloroplast genomes, using total DNA from each species as the templates, and the ensuing products were sequenced by Beijing Tsingke Biological Technology Inc. For all six fragments, Sanger sequencing obtained exactly the same sequences as those in the assembled chloroplast genome.These results provide strong evidence that our assembled chloroplast genomes were robust and error-free.
Chloroplast genome annotation and physical map drawing
The DOGMA tool (Dual Organellar GenoMe Annotator;http://dogma .ccbb.utexa s.edu/) was used to annotate the assembled chloroplast genomes (Wyman et al. 2004). These annotations were rectified using Sequin (v.15.10) and submitted to the NCBI database, where the chloroplast genomes received accession numbers MK552376 (H. rhamnoides subsp. yunnanensis) and MK552375 (H. gyantsensis). Both chloroplast genome sequences were uploaded to OrganellarGenomeDRAW (https://chlor obox.mpimp-golm.mpg.de/OGDra w.html) to clearly label the locations of the IR, LSC,and SSC regions and produce an annotated circular chloroplast genome map (Fig. 1).
Comparative analysis of the two Hippophae chloroplast genomes
DnaSP (v.5.0) was utilized to analyze sequence divergence between H. rhamnoides subsp. yunnanensis and H. gyantsensis chloroplast genomes (Librado and Rozas 2009). When screening for base differences between chloroplast genomes,the sliding-window method was used for the calculations(setting window length to 600 bp and step size to 200 bp in this analysis). Selection pressure on each protein-coding gene was inferred by calculating the ratio of nonsynonymous to synonymous substitutions (i.e., Ka/Ks), using the KaKs_Calculator (v.2.0) (Zhang et al. 2006) and its default parameters.
SSR loci analysis
The SSR loci of the two chloroplast genomes were located using MISA software (http://pgrc.ipk-gater slebe n.de/misa/misa.html). The minimum number of repeat mononucleotide, dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide units was 10, 5, 4, 3, 3, and 3,respectively (Li et al. 2018); the shortest distance between any two SSR loci was set to 100 bp.
Optimal codon analysis
Optimal codon usage in each protein-coding gene was analyzed in both H. rhamnoides subsp. yunnanensis and H.gyantsensis chloroplast genomes. Repetitive sequences in their protein-coding genes were removed, and only genes longer than 300 bp that used ATG as the initiator codon and TAA, TAG, or TGA as the terminator codon were analyzed (Hu et al. 2016; Liu et al. 2017). This yielded 53 gene sequences for the optimal codon analysis from 85 proteincoding genes in each chloroplast genome. Next, the effective number of codons (ENC) value per gene was used as the criterion to sort these 53 gene sequences. Finally, five genes with the lowest ENC values were used to construct highexpression gene pools, and five genes with the highest ENC values were used to construct low-expression gene pools.The software tool CodonW (v.1.4.2) (Xu et al. 2011) was used to obtain the relative synonymous codon utilization(RSCU) value for each codon of the 53 genes, and also the ΔRSCU value for those codons in the high- and low-expression gene pools. Optimal codons were designated as those with RSCU > 1 and ΔRSCU > 0.08 (Wang et al. 2018a, b).
Cluster analysis of chloroplast genomes
Whole chloroplast genomes of three species in the Elaeagnaceae family- Elaeagnus macrophylla (NC_028066),Elaeagnus mollis (NC_036932), and H. rhamnoides(NC_035548.1)-and one in the Rhamnaceae family,Ziziphus jujube (NC_030299), were download from NCBI,then aligned with the two Hippophae chloroplast genomes assembled in this study (using BioEdit v.7.1.3, Ibis Biosciences, Abbott Laboratories, Carlsbad, CA, USA). Phylogenetic trees for all six chloroplast genomes were constructed using maximum likelihood (ML) and maximum parsimony (MP) with 1,000 bootstrap replications and default parameters in MEGA (v.7.0) (Kumar et al. 2016)and using the Bayes cluster method in MrBayes (v.3.1.2)(Huelsenbeck and Ronquist 2001) (parameter settings:Nst = 6; rates = Invgamma; mcmc ngen = 1,000,000; print frequency = 100; sample frequency = 100; number of chains = 4; sumt burnin = 2500.) The chloroplast genome sequence of Z. jujube served as the outgroup in this analysis.
Results
Basic characteristics of the chloroplast genomes

Fig. 1 Physical map of the chloroplast genome of Hippophae gyantsensis and H. rhamnoides subsp. yunnanensis
The full length of the H. gyantsensis chloroplast genome was 155,260 bp, a little shorter than the length of the H.rhamnoides subsp. yunnanensis chloroplast genome(156,415 bp). The length of the LSC, SSC and IR regions were, respectively, 83,026 bp, 18,894 bp, and 26,670 bp in H. gyantsensis, and 84,078 bp, 19,047 bp, and 26,648 bp in H. rhamnoides subsp. yunnanensis. The difference between the two Hippophae chloroplast genome lengths in their protein-coding region was just 5 bp (Table 1). The GC concentrations in the whole chloroplast genome and each of its regions were similar between the two Hippophae species and greatest in the IR region.
Both chloroplast genomes contained 131 functional genes, comprising 85 protein-coding genes, 38 transfer RNA(tRNA) genes, and eight ribosomal RNA (rRNA) genes(Table 2). Among them, four rRNA, eight tRNA, and seven protein-coding genes were located in the IR regions; 13genes were found in the SSC region; and the rest belonged to the LSC region (Fig. 1). Notably, the 5′ end of rps12, a trans-splicing gene, is located in the LSC region, with one copy of its 3′ end in each IR region. Two ycf1 genes found differed in length, with a shorter segment located in one IR region and a longer segment located in the other IR region and the SSC region. There were 22 genes with introns; the clpP, rps12, and ycf3 genes contained two introns, while the rest had one intron. Evidently, the lengths of introns in the two Hippophae chloroplast genomes were similar. The ndhA gene showed the largest difference in intron length between the two species, being 20 bp larger in H. gyantsensis than in H. rhamnoides subsp. yunnanensis; the trnK- UUU gene featured the longest intron, 2485 bp in H. gyantsensis and 2497 bp in H. rhamnoides subsp. yunnanensis (Table 3).

Table 1 Comparison of the complete chloroplast genomes for Hippophae gyantsensis and H. rhamnoides subsp.yunnanensis

Table 2 Chloroplast genome coding information of Hippophae gyantsensis and H. rhamnoides subsp. yunnanensis

Table 3 Characterization analysis of introns in the Hippophae gyantsensis and H.rhamnoides subsp. yunnanensis chloroplast genomes
A total of 100 SSR loci were detected in the H. gyantsensis chloroplast genome, comprising 75, 15, 4, 5, and 1 of one-, two-, three-, four-, and five-base SSR loci, respectively. In H. rhamnoides subsp. yunnanensis, 80 SSR loci were detected consisting of 54, 16, 4, and 6 of one-, two-,three- and four-base SSR loci, respectively. All the one-base SSR loci of both Hippophae species were A/T repeats, and all their two-base SSR loci were AT repeats, except for one CT repeat locus in H. gyantsensis. The SSR loci of the two chloroplast genomes were mainly located in the noncoding region, with just 13 and nine SSR loci found in the coding sequences of H. rhamnoides subsp. yunnanensis and H. gyantsensis , respectively (six of these were at the same genome locations in each species).
Base differences between chloroplast genomes
After comparing the whole chloroplast genomes of the two Hippophae species, we found 288 locations with base sequence insertions/deletions, mainly located in the LSC region, of which 20 exceeded 50 bp. Of 1302 nucleotide substitutions identified, 478 (36.71%) were in the coding region and 824 (63.29%) in the intergenic or intron regions.By comparing 92,450 bp and 62,810 bp lengths, substitution rates in the coding and intergenic or intron regions between complete chloroplast genomes of the two species were estimated at 1.31% and 0.517%, respectively.
According to the sliding-window analysis, the greatest difference between the two Hippophae species lay in the SSC region, followed by the LSC region, with the IR regions the least different (Fig. 2 a). Similar results were obtained for the coding region, in that the longer ycf1 gene segment of the SSC region had the greatest number ofloci with base differences per length, followed by the ndhF gene in the same region; the matK, rps16, rpoC2, ndhK, and ndhC genes in the LSC region had a relatively high number of base different loci per unit (π ≥ 0.15) (Fig. 2 b). By contrast, differences in the IR region were all small (π < 0.05). As Fig. 2 a compared with 2 b shows, the locations with large base differences are usually in the intergenic region.

Fig. 2 Sliding-window analysis of the chloroplast genomes of Hippophae gyantsensis and H. rhamnoides subsp. yunnanensis. X-axes indicate the base location; Y-axes indicate the nucleotide poly-morphism (π value); a base difference of the complete chloroplast genomes and b base difference of gene CDS region (not including introns)
The Ka/Ks analysis suggested 472 substitute loci exist between the two Hippophae species in all 85 protein-coding genes; 264 of them are Ks and 208 of them are Ka,for an overall Ka/Ks ratio of 0.230. Gene sequences of 19 protein-coding genes were identical between the two species, comprising nine self-replicating-related genes and 10 photosynthesis-related genes. There were 63 protein-coding genes with Ka/Ks ratios < 1, of which 46 were Ka/Ks ≪ 1 and statistically significant (p ≤ 0.05). Excluding those genes with only one or two synonymous substitutions, or with only one nonsynonymous substitution, left 25 genes consisting of 10 protein-coding genes related to photosynthesis, 11 protein-coding genes related to self-replication, and four genes with other functions. Although the matK , rps16, and rpoA genes each had values of Ka/Ks > 1, these ratios were not significant (p> 0.05) (Table 4).
Comparison of the optimal codons of the chloroplast genomes
We identified 20 optimal codons in the chloroplast genome of H. gyantsensis and 16 in that of H. rhamnoides subsp.yunnanensis, of which 15 were shared between the two species (Table 5). Regarding the amino acids encoded by these codons, besides tryptophan-which has only one codon and no codon usage preference-only aspartic acid and cysteine in H. gyantsensis, and phenylalanine, aspartic acid, cysteine,and glycine in H. rhamnoides subsp. yunnanensis, lacked any optimal codons. For all optimal codons in the two species, only UUG (coding for leucine) ended with G; since the rest all ended with A or U, this result indicated a significant AT preference.
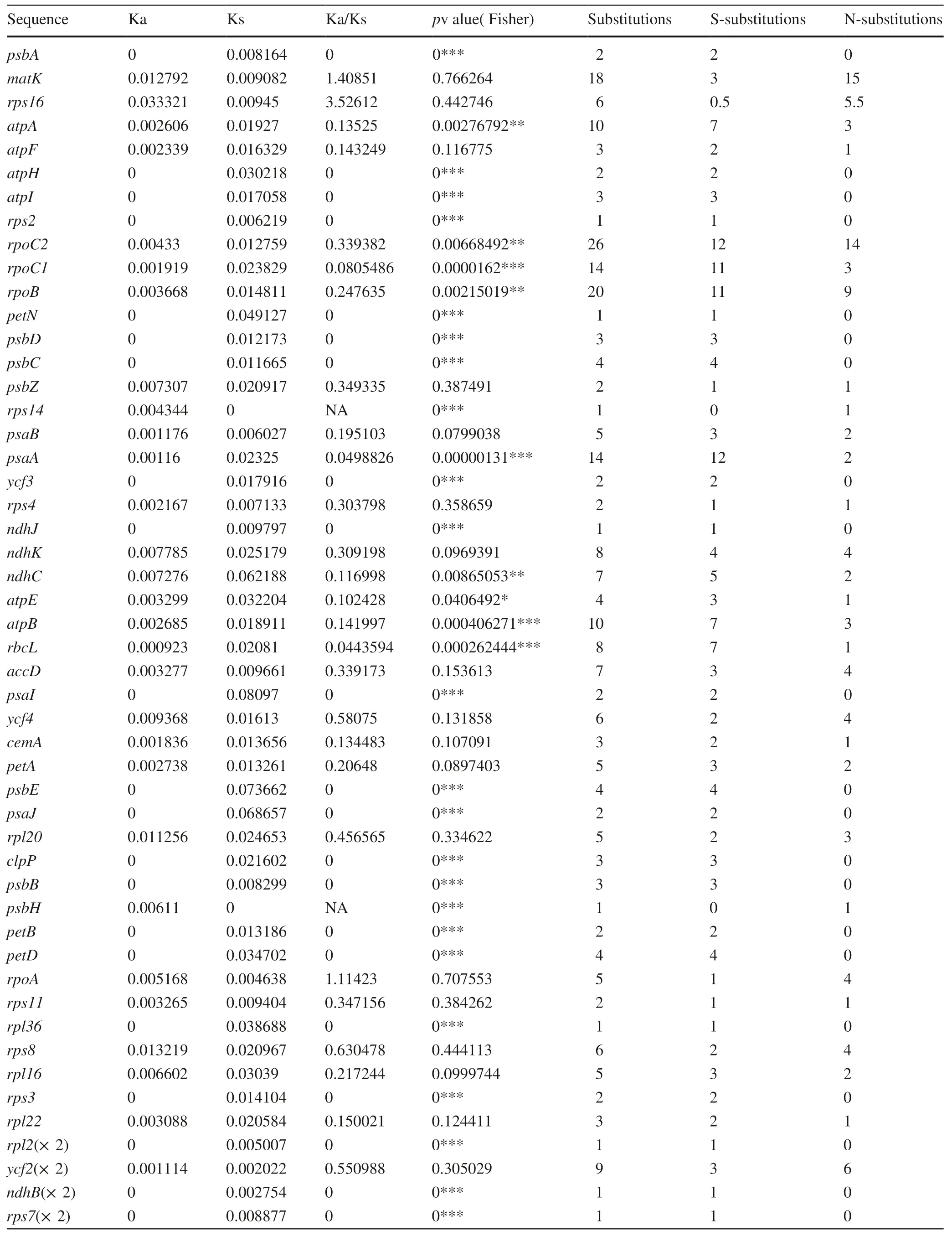
Table 4 Ka/Ks analysis of chloroplast genomes for the CDS region of Hippophae gyantsensis and H. rhamnoides subsp. yunnanensis

Table 4 (continued)
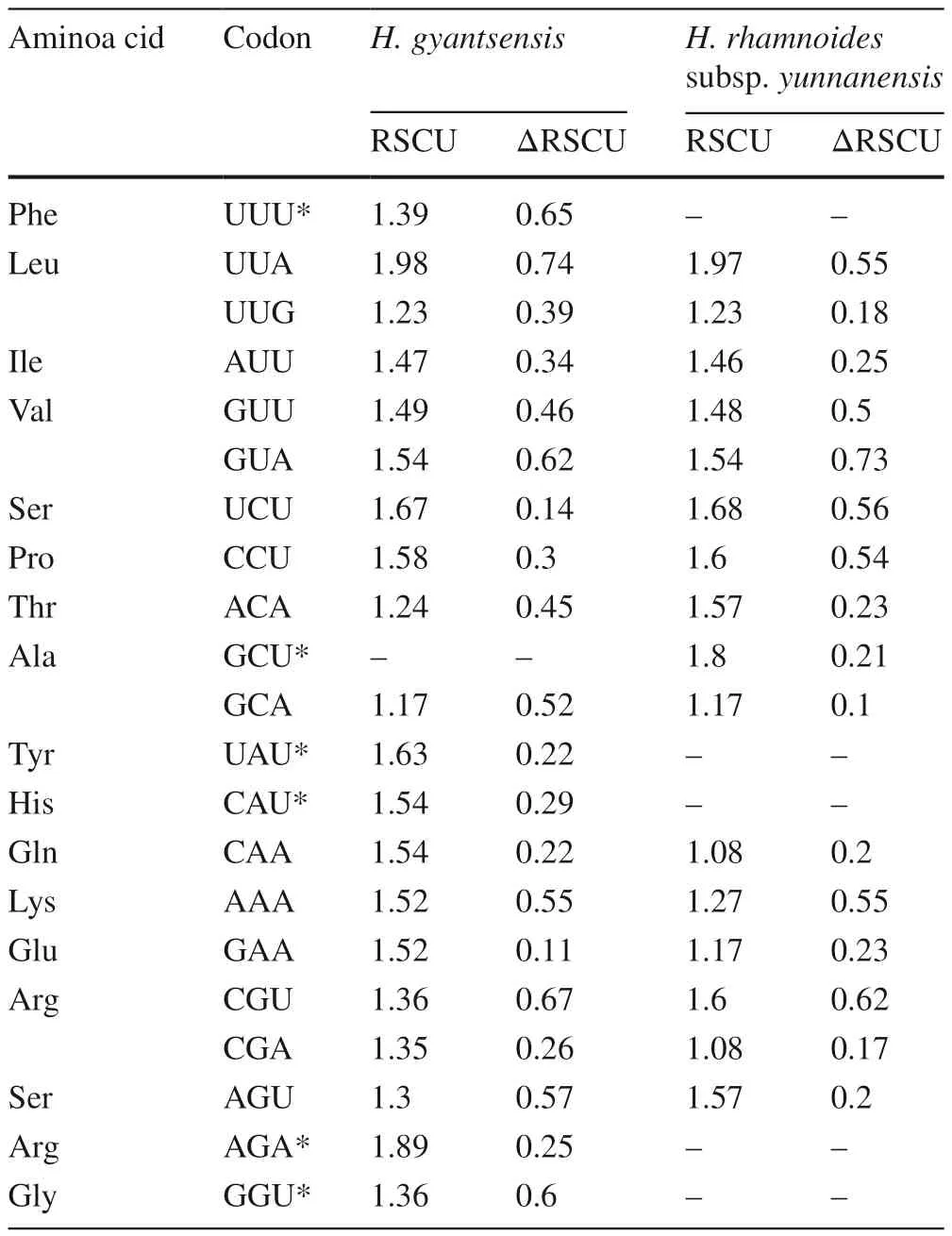
Table 5 Comparison of optimal codons in Hippophae gyantsensis and H. rhamnoides subsp. yunnanensis chloroplast genomes
Phylogenetic relationship of chloroplast genomes
Phylogenetic trees based on BI, ML, and MP methods all suggested that, using one Ziziphus jujube sequence as the outgroup, all five Elaeagnaceae species formed a monophyletic clade with two subclades. Two species from the genus Elaeagnus formed one subclade, while the other comprised three Hippophae species. Within this last subclade, H. rhamnoides subsp. yunnanensis was more closely related to H.rhamnoides than H. gyantsensis. In all three trees, all clades received bootstrap support values of 100% (Fig. 3).
Discussion
Basic characteristics of complete Hippophae chloroplast genomes
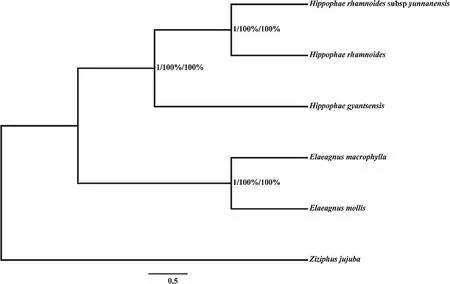
Fig. 3 Cluster analysis of six chloroplast genomes. MrBayes, maximum likelihood and maximum parsimony methods were used for the cluster analysis. The chloroplast genome sequence of Z. jujube served as the outgroup
In this study, we assembled and annotated the whole chloroplast genome sequences of two woody plants, H. gyantsensis and H. rhamnoides subsp. yunnanensis. When compared with several published full-length chloroplast genomes,those of the two Hippophae species were similar to H. rhamnoides (Chen and Zhang 2017), and to two other species in the same family, E. macrophylla (Choi et al. 2015) and E.mollis (Wang et al. 2017), while perhaps also close to Arabidopsis thaliana (Sato et al. 1999) and some alpine plants,such as Pedicularis cheilanthifolia (Zhang et al. 2017) and Orinus thoroldii (Liu et al. 2017). All of these genomes have the typical quadripartite structure of angiosperm chloroplast genomes, with two copies of IR regions separated by SSC and LSC regions. Since the number and type of genes in the two Hippophae chloroplast genomes are quite similar to those of the above comparison species, the chloroplast genome has a highly conserved structure. General purifying selection could explain this conservation for angiosperms over their evolutionary history and is partly supported by the Ka/Ks analysis between the two Hippophae chloroplast genomes, which revealed that nearly half of protein-coding genes were identified as being subjected to significant purification effects (Table 4).
We found 1302 nucleotide substitutions between H.gyantsensis and H. rhamnoides subsp . yunnanensis chloroplast genomes. Most of these (~ 63%) occurred in the intergenic region or intron sequences and the rest (~ 37%)in coding sequences, which represented 1.31% and 0.52%of all their sequences, respectively. The SSC region clearly has the highest substitution rate. The longer segment ycf1,ndhF, and ccsA genes, which had the highest substitution rates, are completely or mostly located in this crucial region.The ycf1 gene is known to be widespread in plant chloroplast genomes (Zhang et al. 2013; Jan et al. 2015; Dong et al.2015, 2018). Not surprisingly then, many studies have used this gene to analyze interspecies genetic diversity in plants due to its high substitution ratio (e.g., Datwyler and Weiblen 2004; Zhao et al. 2016). Nevertheless, a high substitution rate in the ccsA gene between our two Hippophae species conflicts with an earlier conclusion that this gene is widely conserved in photosynthetic plants (Wicke et al. 2011). The LSC region had the second highest substitution rate, wherein matK, rps16, rpoC2, ndhC, and ndhK genes showed relatively high substitution rates in both Hippophae species. By contrast, their IR regions were highly conserved, which is consistent with findings for many other angiosperm species(Ruhlman and Jansen 2014; Li et al. 2016; Dong et al. 2018;Meng et al. 2018).
Many factors can influence genomic substitution rates.Following a mutation event, a plant chloroplast could maintain the integrity of its genetic material by multiple processes. Of particular importance among these processes is the extensive use of DNA recombination (Maréchal and Brisson 2010). In the chloroplast genome of most flowering plants, a frequent and easily observable flip-flop recombination event occurs between the two IR regions (Maréchal and Brisson 2010), which would lead to differential substitution rates between the single-copy regions and the IR region, suggesting an efficient gene conversion mechanism is operating. Such a mechanism has been invoked to explain recombination copy correction in chloroplast transformants when point mutations or foreign sequence are introduced into the IR regions (Wicke et al. 2011; Ruhlman and Jansen 2014). This factor therefore may be a critical contributor to chloroplast stabilization.
Optimal codons characteristics of Hippophae
In codon usage bias analysis, now a key tool for exploring genetic and evolutionary pathways of species (Zhao et al.2016), optimal codons can provide an important representation of codon usage bias. Here we found 15 common optimal codons for H. rhamnoides subsp. yunnanensis and H. gyantsensis chloroplast genomes, which reflects the high commonality of codons in Hippophae plants. That all but one codon (ending with G) ended with A or U is evidence for a significant AT preference, which is consistent with many findings on optimal codons in angiosperms (Hershberg and Petrov 2009; Hu et al. 2016). Directional mutation and natural selection are two main factors that drive codon-usage bias(Wang et al. 2018a, b). Under directional mutation, the utilization of certain optimal codons is matched with the nucleic acid content in the chloroplast genome’s intergenic region(Hershberg and Petrov 2009). Usually, selective action does not affect the bases in the intergenic region, so high AT content there probably occurs because those bases are more likely to mutate into A/T. We estimated the AT content in the intergenic regions of both Hippophae chloroplast genomes is 67.4%, which is higher than that in gene regions, indicating that directional mutation is likely the principal factor influencing codon-usage bias in Hippophae chloroplast genomes.Of course, natural selection may also have important effects and merits further study.
Conclusions
Through comparison and analysis of chloroplast genome sequences of H. rhamnoides subsp. yunnanensis and H.gyantsensis, we showed that the length of the Hippophae chloroplast genome is consistent with most model plants and has the typical four-section structure of angiosperm chloroplast genomes. The SSC region has the highest base substitution rate, whereas the IR regions are highly conserved. Hippophae species may have a high commonality of codons. These conclusions provide a valuable reference for future studies on Hippophae plants, including those on the characteristics of chloroplast genomes of angiosperms in high-elevation areas.
杂志排行

Journal of Forestry Research的其它文章
- A commentary review on the use of normalized difference vegetation index (NDVI) in the era of popular remote sensing
- Reconciliation of research on forest carbon sequestration and water conservation
- A theory to link relationships of stand volume, density, mean diameter and height in forestry data
- A new model for predicting the total tree height for stems cut-to-length by harvesters in Pinus radiata plantations
- Comparative performances of new and existing indices of crown asymmetry: an evaluation using tall trees of Eucalyptus pilularis(Smith)
- Tree mortality and biomass loss in drought-affected forests of East Texas, USA