Phylogenetic relationship of Picea mongolica with other Picea species in the same area based on chloroplast gene variations
2021-01-11HaoDiJingjingMaKuaikuaiHeFangxuHanYueLiShihuiNiu
Hao Di · Jingjing Ma · Kuaikuai He · Fangxu Han ·Yue Li · Shihui Niu
Abstract Picea mongolica is a conifer with a limited distribution in China and its taxonomic status is controversial. In order to explore genetic differences between P.mongolica and other nearby Picea species and to investigate its taxonomic status, phylogenetic relationships were analyzed between P. mongolica and Picea koraiensis,Picea meyeri and Picea wilsonii by three chloroplast gene sequences matK, chlB and atpA. The length of joint chloroplast sequence is 2379 bp. The fifteen haplotypes were identified by haplotype network analysis, among which two were major haplotypes and nine were unique. In addition,the genetic diversity of the sample collection species was inferred. Based on the haplotype network and Neighbor Joining phylogenetic tree analysis, P. mongolica was located on the basal clade of the phylogenetic tree which had more primitive taxa, and the genetic diversity of P. mongolica was highest. The significant differences between P. mongolica and these other Picea species were identified by this research.
Keywords cpDNA · Genetic difference · Phylogeny ·Picea mongolica · Taxonomic status
Introduction
There are approximately 40 species of Picea distributed in the northern hemisphere (Xue et al. 2004), and 16 of these species and 9 varieties occur in China (Zheng 1978). Picea mongolica (H.Q.Wu) W.D. Xu was identified as a new species in 1994 by Xu et al. (1994) according to morphological examination and isozyme and karyotype analysis. Its natural distribution is in a narrow region of sandy soils in eastern Inner Mongolia (43° 30′-43° 36′ N, 117° 06′-117°16′ E), forming rare sandy forest grasslands (Xu 1993; Xu and Zheng 1993). P. mongolica is an excellent species for afforestation on the sandy soils of this region compared with other species (Xu and Zou 1998).
Due to its unique biological characteristics and limited geographical distribution, it has attracted considerable attention. The physiology and ecology of P. mongolica has been studied extensively (Zou 2004). However, its taxonomic status has been controversial. Some researchers consider that it is a variety of P. meyeri (Zhu 1958), located in the south of the P. mongolica region), and others believe that it is Picea koraiensis (Zheng 1978, 1983) found in northern China. Wu (1986) suggested that P. mongolica could be a land race of Piciea meyeri (P. meyeri var. mongolica) based on multivariate statistical analysis of anatomical features and morphological traits, and this theory was later supported by Li et al. (2008). The debate on whether P. mongolica should be a distinct taxonomical species has lasted for decades. Previous studies have focused on morphology with little or no attention to molecular aspects. Research which analyzed morphological characteristics by RAPD markers of pollen (Cai et al. 2009; Zhang 2009) considered that P.mongolica should be an independent species. A report based on ISSR markers supported the independent species status of P. mongolica as well (Zheng et al. 2015), as there are large genetic differences between P. mongolica and P. koraiensis or P. meyeri.
Because of fairly close phylogenetic relatives and unstable analogous morphological characteristics of Picea, the morphological evidence is not strong enough for classification (Li et al. 2011). DNA sequences based on molecular markers can provide stronger evidence in the genetic differentiation of species. Research shows that molecular markers with high interspecific gene flows may prevent intraspecific introgression (Nybom 2010; Gautam et al. 2018), and theoretically molecular markers are more applicable for species identification (Li et al. 2008). In the Pinaceae, chloroplast genomes are mostly paternally inherited and transmitted through the pollen grains. Compared with nuclear and mitochondrial genomes, chloroplast genomes have higher gene flow which is more suitable for the def inition of species(Petit et al. 1993, 2005; Du et al. 2009).
The application of cpDNA in phylogenetic studies of Picea is common but has not been reported for P. mongolica. In this study, chloroplast DNA (cpDNA) was used to investigate the genetic diversity and phylogenetic status of P. mongolica. Sampling covered most of this species range in China (Table 1). Two cpDNA sequences were selected with faster evolution rates-MatK and chlB, and one with a slower evolution rate, atpA, to study genetic differences of P. mongolica from other nearby species, P. koraiensis, P.meyeri and P. wilsonii (Jia 2011). We focused on the genetic structure and possible phylogenetic relationship of P. mongolica. Four major issues were addressed: (1) whether there are mutations in the chloroplast genes of P. mongolica and can they be used to def ine this species; (2) the analysis of genetic diversity of chloroplast genes between different species and their spatial patterns; (3) to establish the phylogeny of P. mongolica based on chloroplast gene mutations; and,(4) whether P. mongolica should be a taxonomical species.
Materials and methods
Plant materials and DNA extraction
Plant materials were collected from three populations covering most of the P. mongolica distribution in Inner Mongolia.Three other species in the same area, P. koraiensis, P. meyeri, P. wilsonii were included for genetic analysis (Fig. 1).Due to the occurrence of forest fires, the spread of pests and diseases, and poor management, older trees were selected at intervals of 50 m to avoid collecting genetically identical individuals. Forty individuals were sampled from each species. Latitude and the longitude of each population are recorded in Table 2. A total of 160 individuals were used in this study. Fresh needles were dried with silica gel and stored at − 20 °C. Total genomic DNA was extracted from needles using a Plant Genomic DNA Kit (Tiangen Biotech,Beijing, China). The isolated DNA was dissolved in TE buffer and stored at − 20 °C before use.
DNA amplification and sequencing

Fig. 1 Geographical distribution of P. mongolica with its nearby species

Table 1 Locations of samples and haplotype distribution
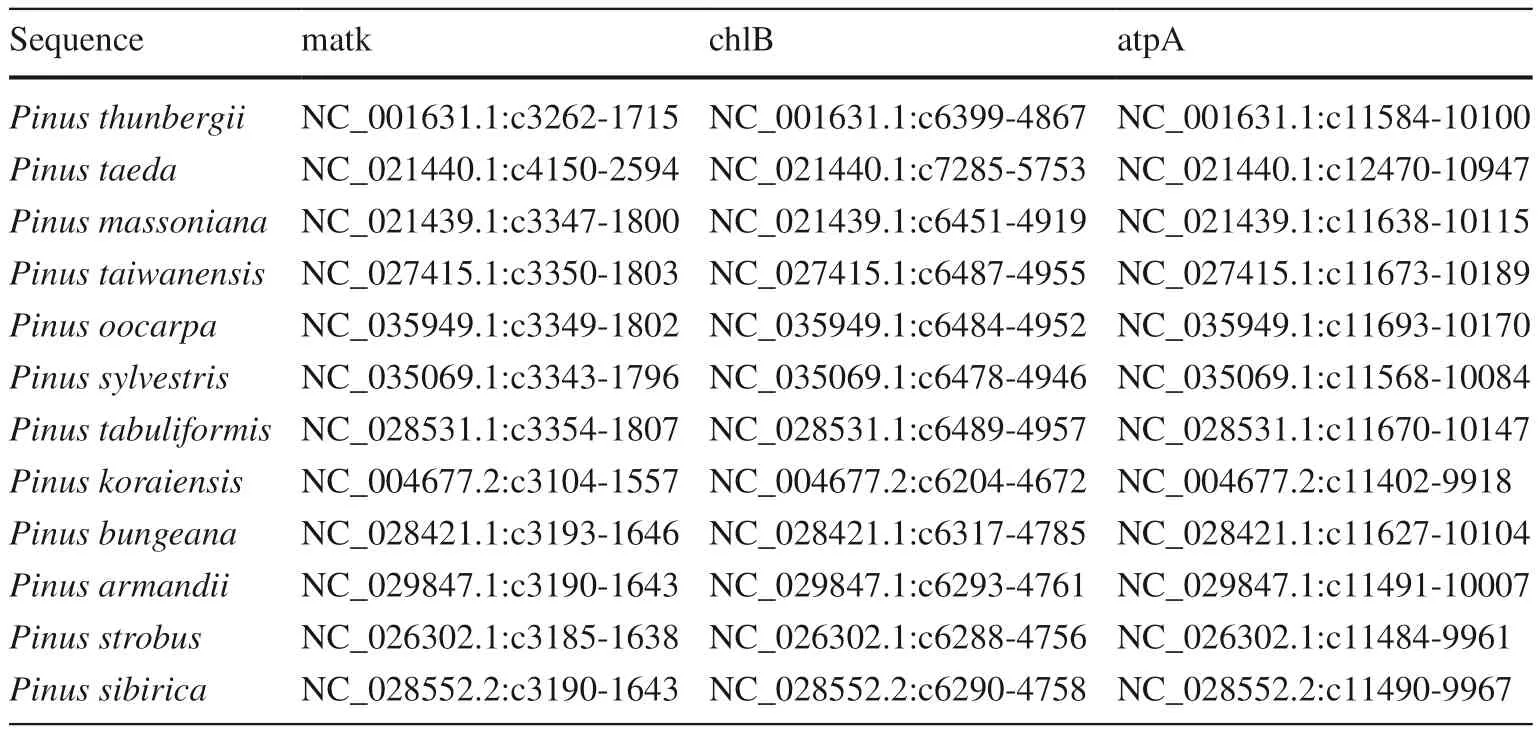
Table 2 Species name and sequences information of Pinus
Gene sequences of three cpDNA regions (matK, chlB, atpA)were obtained in this study (Table 2). The primers of amplification and sequencing were designed by BatchPrimer3 v1.0 (https ://wheat .pw.usda.gov/demos/Batch Prime r3/).The original sequences for primers were obtained from Picea glauca in National Center for Biotechnology Information (NCBI) (matK: NC_028594.1:c3297-1750, chlB:NC_028594.1:c6248-4707, atpA: NC_028594.1:49700-51223). Information on the primers is shown in Table 3.Standard polymerase chain reaction (PCR) amplification was performed in 25 μL reaction systems consisting of 22 μL 1 ×PCR Mix (TSINGKE, Beijing, China), 1 mmol/L each primers, 30 ng DNA template. The program was set as an initial denaturation of 2 min at 98 °C, followed by 35 cycles of 10 s at 98 °C, 15 min annealing at 55 °C, and 20 s extension at 72 °C, with a final extension of 2 min at 72 °C. PCR product sequencing was carried out by the biological company(TSINGKE, Beijing, China). In addition, matK, atpA and chlB gene sequences of Selaginella moellendorffii (matK:NC_013086.1:32629-34227, atpA: NC_013086.1:23686-25206, chlB: NC_013086.1:29063-30604) and 12 Pinus species were obtained from NCBI for the analyses of the phylogenetic evolution of Picea (Table 2).
Date analysis
Sequences were aligned by CLUSTAL × 2.0 (Thompson et al. 1997; Larkin et al. 2007) and adjusted manually with BioEdit version 7.0.0 (Hall 1999). They were then concatenated into one matrix. The haplotypes of cpDNA were determined based on nucleotide substitutions and indels with DnaSP (version 5.10.01; Librado and Rozas 2009).Haplotype diversity (Hd), nucleotide diversity (π) (Nei and Tajima 1983), and Tajima’s D (Tajima 1989) were also calculated in DnaSP5 (Rozas et al. 2003). Analyses of molecular variance (AMOVA) (Excoffier et al. 1992) were performed to assess genetic differentiation within populations,among populations within species and among species using the program Arlequin version 3.0 (Excoffier et al. 2005).Population genetic differentiation (Fst) (Weir and Cockerham 1984) was also computed. The phylogenetic tree based on genetic distance and haplotypes were both constructed by the neighbour-joining (NJ) method in MEGA5 with 1000 bootstrap replications (Tamura et al. 2011). In addition, a network for the cpDNA haplotypes was constructed in R(https ://arund urvas ula.wordp ress.com/2016/02/24/haplo type-netwo rks-in-r).
Results
Differences of chloroplast genes between P. mongolica and its nearby spruce species
After alignment and manual adjustment, the final fragment length matK, chlB and atpA were 699, 863 and 816 bp,respectively. Two polymorphic sites were detected in the atpA sequence, and the nucleotide diversity was 0.00048.The atpA had three haplotypes in four species, and the haplotype diversity was 0.383. In the chlB sequence, onepolymorphic site and two haplotypes were detected, and the nucleotide diversity and haplotype diversity were 0.000061 and 0.052 respectively. The matK sequence with 7 polymorphic sites and 10 haplotypes had the most haplotypes. The diversity of the nucleotide was 0.00078, and the haplotype diversity 0.379 (Table 4). Compared with the three chloroplast gene sequences, the variation range of nucleotide diversity was from 0.000061 to 0.00078, and the order ranged from large to small, matK to atpA, chlB. The haplotype diversity was from 0.052 to 0.383, and the order of several to a few was atpA to matK, chlB.
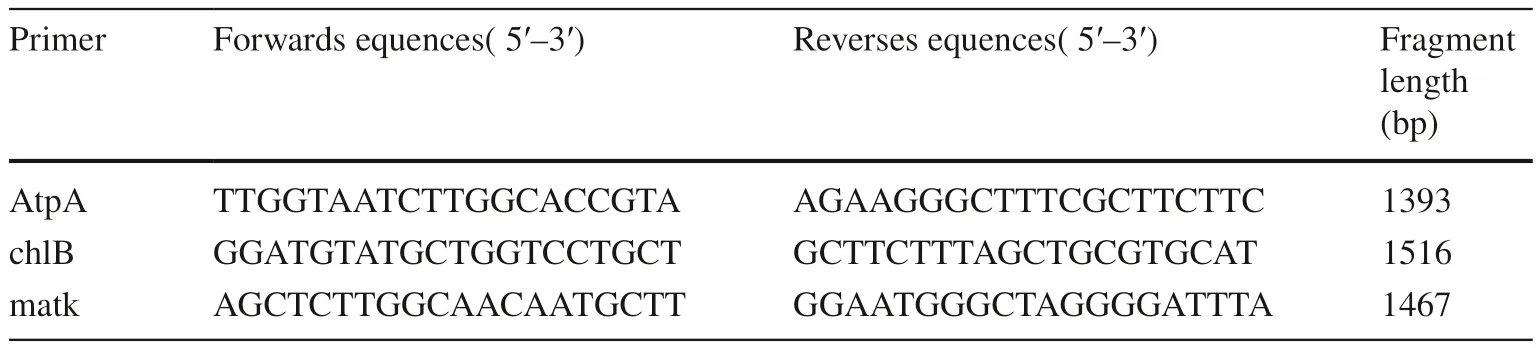
Table 3 PCR amplificating Primers of cpDNA
The total length of the concatenated alignments was 2379 bp. Because of failure during gene amplifying and sequencing, 101 joint cpDNA sequences were eventually obtained. Ten variation sites were identified, including seven nucleotide substitutions and three indels (Table 5).The diversity of nucleotide was 0.00043, and the diversity of haplotype 0.652 (Table 4). Based on these variations, 15 haplotypes were identified. The distribution of haplotypes was shown in Tables 2 and 6. Among the 15 haplotypes,two (Hap 14, Hap 15) were major shared between four species. In addition, Hap 9, Hap 10, Hap 11 and Hap 12 occurred in two species. In contrast, Hap 1 to Hap 8 and Hap 13 were found in only one population, and thus were considered unique haplotypes. These accounted for 53% of all haplotypes, more than the number of common haplotypes.P. mongolica had the most haplotypes, a total of 13, seven of which were only found in P. mongolica. P. wilsonii had seven haplotypes, including two unique haplotypes. There were no unique haplotypes in P. koraiensis and P. meyeri.
Genetic diversity and differentiation of P. mongolica
Nucleotide and haplotype diversities and Tajima’s D of six populations from Picea are summarized in Table 7. Nucleotide diversity (π) within each population ranged from 0.0001 to 0.0008, while haplotype diversity (Hd) varied between 0.3 and 0.9. With the same nucleotide diversity, the highestlevel of genetic diversity was detected in the SD2 population(π = 0.00087, Hd = 0.96), and population B (π = 0.00014,Hd = 0.32) had the lowest haplotype and nucleotide diversities. Interestingly, the three populations with the highest haplotype and nucleotide diversities were P. mongolica . An analysis of molecular variance (AMOVA) showed that the majority of variations occurred within populations (84.2%;Table 7). Variation existing among Picea populations was 11.3%, while variations within species was only 4.4%. The results of Tajima’s D test indicated that there was no significant deviation from neutrality in all populations. The results of population differentiation and analyzes of molecular variance were shown in Tables 8 and 9. Population differentiation (Fst) analysis showed obvious differentiation among populations. This was especially prominent between P.mongolica and its closely related nearby species.
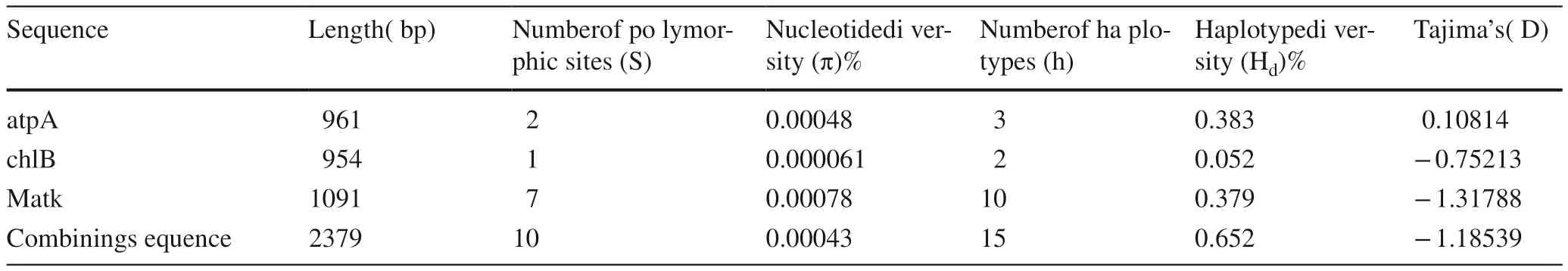
Table 4 Variation and haplotype statistic of cpDNA sequences
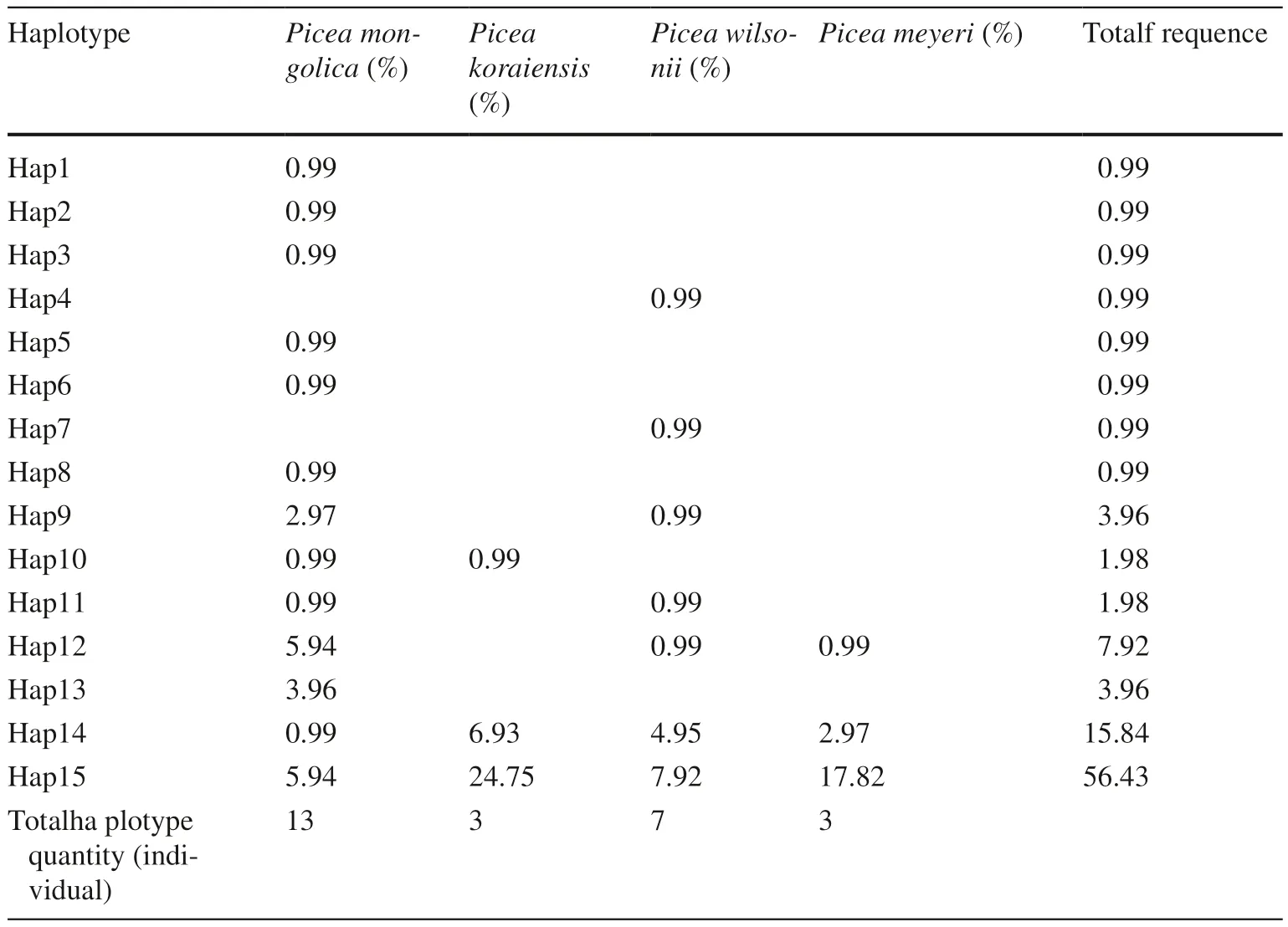
Table 6 Haplotype frequency in four Picea species
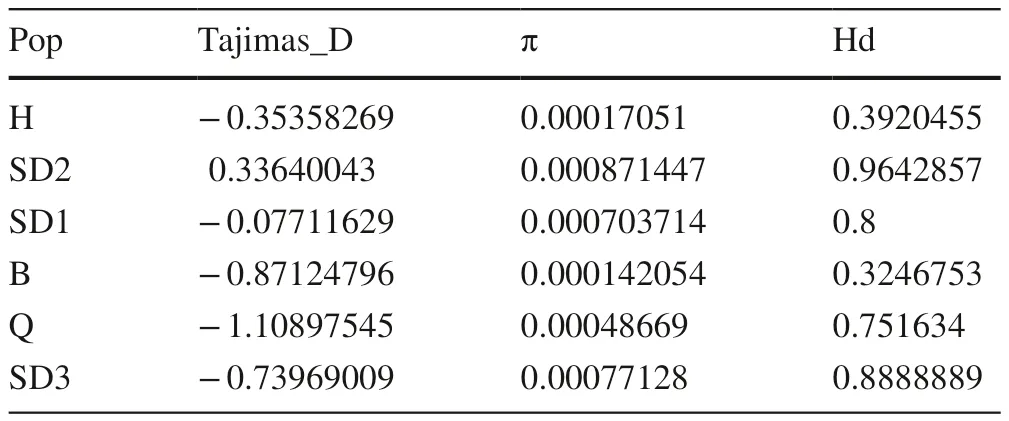
Table 7 Combining sequence diversity index in four Picea species
Phylogeny of P. mongolica based on chloroplast gene mutation
A maximum parsiony tree (Fig. 2) was oriented with divergent combined chloroplast gene fragments and revealed two principal monophyletic lineages (clades).The three groups of P. mongolica were clearly within oneclade. Three closely species, P. koraiensis, P. meyeri, and P. wilsonii, were located in the second clade. This also shows that P. mongolica diverged earlier and belongs to a more primitive taxa, while there is a certain degree of differentiation between different groups of P. mongolica.In addition, the relationship between P. koraiensis, P.meyeri and P. wilsonii is relatively close. However, compared with P. koraiensis and P. meyeri , the relationship between P. mongolica and P. wilsonii is closer. The obvious genetic difference between P. mongolica and the other Picea species suggests that P. mongolica should be an independent species taxonomically.

Table 8 Analysis of molecular variance (AMOVA) of joint cpDNA in four Picea species

Table 9 Population differentiation (Fst) between six populations based on joint cpDNA
The taxonomic status of P. mongolica based on haplotype analyses
In the haplotype NJ tree of the phylogenetic analyse, with Hap 13 as the dividing line, two distinct clades were identified (Fig. 3). The bootstrap value ranged from 7 to 75%.Hap 3, Hap 7, Hap 9, Hap14, Hap10, Hap15, Hap 6, and Hap 11 consisted of a mixed branch containing four species. Seven haplotypes (Hap 1, Hap 2, Hap 4, Hap 5, Hap 8, Hap 12, and Hap 13) formed a strongly supported clade,including P. mongolica . Among all of the haplotypes, Hap 6 and Hap 11 have the earliest divergence time and are the most primitive haplotypes. Hap 11 includes P. mongolica and P. wilsonii. Hap 6 only includes P. mongolica, both are regarded as ancestral haplotypes. Moreover, this further illustrates the close relationship between P. mongolica and P. wilsonii. In additional, Hap 13 formed another independent subclade. According to the phylogenetic tree branches,it can be further inferred that there is a genetic difference between P. mongolica and other neighbouring Picea species.The network of 15 cpDNA haplotypes was consistent with the topology revealed by phylogenetic analyses (Fig. 4). Hap 14 and Hap 15 were shared between all four species. Hap15 covering 56% individuals is the most widespread haplotype,located in the middle of the network and may have played an important role in the evolution of haplotypes. Based onthe fan area in Fig. 4, P. koraiensis and P. meyeri have less haplotypes, while distribution in the network diagram is relatively concentrated. Conversely, P. mongolica covered more and rarer haplotypes but distribution in the network diagram is relatively scattered and the occupied area relatively small.

Fig. 3 NJ tree of joint cpDNA haplotypes

Fig. 4 Network of joint cpDNA haplotypes. Each circle represents a haplotype with different colors representing different species. The area of the sector represents the proportion of the haplotype that the species contains; the line between different circles represents a possible evolutionary relationship. B is P. meyeri, H is P. koraiensis, Q is P. wilsonii and SD is P. mongolica
Discussion
According to the theory of evolution, the adjacent natural populations may have a closed kinship within a widely distributed species (Gao et al. 2018), so that interspecific differences and the relationship between species could be well identified. There is a relatively large, continuous natural distribution of P. mongolica in the Hunshandake Desert of Inner Mongolia (about 20 km2), while other nearby natural
populations of Picea are relatively small. Therefore, three sampling groups 1000 m apart in the P. mongolica population were selected in order to reflect the genetic features of a large population; a uniform sample distribution of other nearby Picea species was used in this study. Although only 40 samples of P. mongolics were collected, this does not significantly affect the estimation of genetic diversity and variation as the number of samples is greater than 30 (Nei and Tajima 1983; Ru et al. 2018). The sample strategy should reflect the genetic structural characteristics and the differences between adjacent natural populations of each species.
There are mutations in several chloroplast genes of P.mongolica and the type and frequency of haplotypes were significantly different between species. Therefore, the selected chloroplast genes can be used in the phylogenetic analysis of P. mongolica. In the four species, matK and atpA sequences have high polymorphism and abundant haplotypes. Therefore, these two sequences can be preferentially selected in phylogenetic research.
Picea mongolica is an economically and ecologically important conifer in China. In contrast to its limited range,relatively high levels of nucleotide diversity and haplotype diversity were detected which are comparable to the other related species, P. koraiensis, P. meyeri and P. wilsonii. This is consistent with previous studies (Cai et al. 2009). The results of the analysis of molecular variance (AMOVA),showed that the majority of the variation was within the population. The results of Fst, the population genetic variation, indicated a significant genetic differentiation among the populations. In particular, the degree of differentiation between P. mongolica and other nearby Picea species is the largest. In the long-term evolution of species, genetic variation of the population is affected by environmental and historical events and is preserved in the genetic structure of the population (Ru et al. 2018). The chloroplast genomes of Picea are paternal inheritance transmitted by pollen and have a larger gene flow compared to the mitochondrial gene transmitted through the seed (Wang et al. 2011). The differentiation between P. mongolica and the nearby Picea species was obvious, indicating that the gene exchange among the populations was limited. This may be caused by the special geographical structure of northern China (Mao and Wang 2011). The terrain on the southeastern edge of the Hunshandake Desert is higher than the centre and the south is higher than the north. The landform is low hills, piled dunes and other types (Fu 2012). Geographical isolation may play an important role in the differentiation of P. mongolica (Liu et al. 2011). The original randomly distributed alleles were chosen to adapt to different local environments, resulting in differences among groups (Mayr 1954; Grant 1981; Coyne 1992; Orr and Presgraves 2000).
Phylogenetic analyses have made significant contributions to the understanding of species history and speciation(Avise 2000; Niu et al. 2013; Tougard et al. 2013). Based on cpDNA sequence data, the analysis revealed the formation of populations and the relationship of species by gene trees and networks (Sun et al. 2019). The phylogenetic tree shows that there is a certain degree of differentiation between different groups of P. mongolica. In addition, the relationship between P.koraiensis, P. meyeri and P. wilsonii is relatively close. However, compared with P. koraiensis and P. meyeri, the relationship between the P. mongolica and P. wilsonii is closer. For 15 haplotypes, only two were fixed in all species populations. We deduced that P. mongolica might have experienced an extensive bottleneck or founder effects(Wang et al. 2011; Gao et al. 2012). That means the expansion of the P. mongolica group may be restricted by natural conditions. The gene frequency of a few individuals determines the effect of gene frequency in their offspring, which is an extreme genetic drift. In small populations, genetic variation loss because of bottleneck effect, inbreeding, and founder effect.
Wide-spread haplotypes tend to be primitive, and Hap 14 and Hap 15 may be primitive. As can be seen from the phylogenetic tree (Fig. 3) and Table 2, P. mongolica has a number of primitive haplotypes. We speculate that P. mongolica may be a diffusion centre of haplotypes, with Hap 13 located in the middle of the network. It may have played an important role in the evolution of haplotypes. Haplotype polymorphism of P. mongolica was the highest. More primitive populations often have more haplotypes and become a refuge for species under unfavourable conditions. In terms of geographic distribution, P. mongolica is concentrated in the Baiyinaobao Natural Reserve, P. koraiensis in the Xiaoxinganling, Changbai and Jilin mountains in the northern part of Baiyinaobao, and P.meyeri and P. wilsonii in Shanxi and Hebei, respectively, in the southern part of Baiyinaobao(Zheng 1978). Therefore, P. mongolica is located in the middle of several species of Picea and this further supports the possibility that P. mongolica populations may be a potential differentiation zone. According to the haplotype distribution, P. mongolica may have experienced a slow population expansion. The natural distribution of the species and sampling locations are shown in Fig. 1.
The haplotype tree shows 15 haplotypes gathered into two groups. One is a mixed group which includes the four species. Another group is mainly composed of P. mongolica haplotypes. These findings suggest that allopatric fragmentation and discontinuous range expansion might have promoted intraspecific divergence and differentiation. Due to the narrow, limited and non-overlapping distribution of some haplotypes, genetic structure was strong in P. mongolica.According to haplotype diversity and distribution, it was concluded that P. mongolica might be the oldest of the four species in this study, which is inconsistent with the results of previous studies (Yi 2017). The genetic difference between P. mongolica and the other species is obvious. In combination with the results of the evolutionary tree and network,we believe that Hap 13 may play a transitional role during the evolution of the haplotype. The analysis of molecular variance (AMOVA) indicated that variation mainly occurred within populations. The haplotype distribution showed that the unique haplotypes of P. mongolica were the most common but the shared haplotypes among the populations were less frequent. This might be caused by geographical isolation restricting genetic exchange among populations. The P.mongolica ecosystem may have formed about ten thousand years ago. Before 1949, the area of P. mongolica forest was over 80 km 2 . However, due to disease, pests, forest fires and human activities, the present area is only 19.5 km 2 , located in the ecological ecotone and grasslands, the Daxinganling Mountains and Hunshandake Desert transition zone. It lies between agricultural and pastoral regions, and natural factors and human activities have a certain influence on its distribution and expansion (Xu 1993).
Hybridizations and introgressions are frequent between intraspecific lineages and between different species. Most phylogenetic studies have used chloroplast DNA to study the evolution of species (Avise 2000). In this analysis of chloroplast genes, the difference between P. mongolica and the other Picea species was obvious. P. mongolica is located in the basic position of the branch, belonging to the first group of divergence (Fig. 1). The cpDNA haplotype network showed that haplotypes and unique haplotypes of P. mongolica were both the most abundant. The difference between P. mongolica and nearby Picea species is obvious and it is reasonable to assume that P. mongolica should be a distinct taxonomical species. With the highest genetic diversity, P.mongolica may be the ancestor of P. koraiensis, P. meyeri,and P. wilsonii.
Conclusions
This research provides a theoretical reference for the study of the phylogenetic evolution of P. mongolica and Picea species. The data did not reveal a relationship between P.koraiensis, P. meyeri, and P. wilsonii. These studies are based solely on chloroplast gene sequences. Accordingly,that more data are needed to resolve these relationships.The history of speciation of P. mongolica and the historical expansion of the population require a more systematic study of the combination of mitochondrial and nuclear gene information.
AcknowledgementsThis work was supported by “the Fundamental Research Funds for the Central Universities (NO. 2015ZCQ-SW-02)”and the National Natural Science Foundation of China (31870651).
杂志排行

Journal of Forestry Research的其它文章
- A commentary review on the use of normalized difference vegetation index (NDVI) in the era of popular remote sensing
- Reconciliation of research on forest carbon sequestration and water conservation
- A theory to link relationships of stand volume, density, mean diameter and height in forestry data
- A new model for predicting the total tree height for stems cut-to-length by harvesters in Pinus radiata plantations
- Comparative performances of new and existing indices of crown asymmetry: an evaluation using tall trees of Eucalyptus pilularis(Smith)
- Tree mortality and biomass loss in drought-affected forests of East Texas, USA