用于含芳烃废水处理的室温凝胶剂及其机理研究
2021-01-11赵君彦张宝浩
宋 健,阮 凯,赵君彦,张宝浩,张 宝
(1. 天津大学化工学院,天津 300350;2. 天津市津南区环境监测中心,天津 300350)
超分子凝胶是指由低分子量有机凝胶剂在溶剂中自组装成三维网状结构,并通过界面张力和毛细作用束缚溶剂形成的一类准固态材料[1-2].由于非共价键相互作用在一定条件下是可逆的,因此超分子凝胶具有热可逆性、易加工、自修复性以及刺激响应性等独特的性质[3-4].超分子凝胶在诸多领域得到了广泛的关注,如在溢油处理[5]、纳米材料[6-8]、光电开关[9]、药物释放[10]等领域已有大量研究报道.
随着工业废水泄漏造成的水污染事件的频繁发生,水中有机污染物的去除和回收受到了人们的关注[11-12].例如,2016 年常州市化工厂工业污水外排导致大量氯苯流入地下水中,致使641 名学生换上淋巴癌、白血病等疾病.如何将有毒有机液体(如氯苯、甲苯等)高效环保地从两相混合物中分离出来是一个巨大的挑战[13],目前,主要采用的材料和技术主要包括化学分散剂[14-15]、吸附剂[16-18]、生物降解[19].但是,以上所有材料和技术在实际应用中都存在一定的缺陷.例如,分散剂具有一定的毒性;吸附剂虽然吸附效率高,但选择性较差且吸收的有机溶剂含水,难以回收利用;生物降解存在速度慢、有一定的安全隐患.
目前,对有毒有机液体相选择凝胶化的研究已经成为热点[20-22].自2001 年Bhattacharya 等[23]首次报道了基于氨基酸衍生物的相选择性超分子有机凝胶剂(PSOGs)以来,已经开发出许多不同化学结构的PSOGs,如氨基酸类[24-25]、糖类[26-27]、有机盐[28]等.但目前已报道的PSOGs 大多都需要使用毒性大的助溶剂来实现相选择凝胶,限制了实际的应用.因此,能够在室温条件下直接以粉末形式凝胶有毒有机液体的PSOGs 是更好的水污染处理材料.文献[29-32]报道了一种亮氨酸衍生物,能够以粉末形式在室温下使原油凝胶化,并提出了一种使用乙腈润湿凝胶剂的方案,可以大幅度提高不同类型有机凝胶剂的凝胶能力.Zhang 等[33]报道了一种以葡萄糖为基础的PSOGs,它可以在室温下简单摇动1 min 后以粉末形式从被污染的水中凝胶化苯胺或硝基苯.这些开创性的工作证明了粉末型PSOGs 潜在的应用价值.尽管在这一领域取得了很大的研究进展,但目前,对氯苯、甲苯等有毒有机溶剂的室温相选择凝胶化的研究还很有限,室温凝胶的机理也有待进一步研究.因此,开发出适用于氯苯、甲苯等芳烃溶剂的新型环保PSOGs,并进一步探究PSOGs 结构与室温凝胶性能的关系是非常必要的.
本文在前期工作的基础上[4],对2,4-(3,4-二氯苯亚甲基)-D-葡萄糖酸酰十六胺(凝胶剂G16)的自组装条件进行调控,降低凝胶剂的结晶性,减小组装体之间的范德华作用力.20 ℃下从甲醇中重结晶制备了相选择凝胶剂2,4-(3,4-二氯苯亚甲基)-D-葡萄糖酸酰十六胺-20 ℃-甲醇(G16-20-Me).该凝胶剂具有室温凝胶性能,能在室温下以粉末形式使氯苯、甲苯等芳烃溶剂凝胶化.通过对室温凝胶机理的研究,为更好地理解室温凝胶现象并制备室温凝胶剂提供了一些策略.此外,芳烃的回收实验表明该凝胶剂在含芳烃废水处理领域具有潜在的应用价值.
1 实 验
1.1 实验原料及仪器
3,4-二氯苯亚甲基-D-葡萄糖酸甲酯按照前期本课题组的合成方法制备[4].十六胺、催化剂4-二甲氨基吡啶(DMAP)以及各种溶剂均直接购买自阿拉丁试剂(上海)有限公司.红外光谱数据使用FTS3000光谱仪进行采集.针对凝胶剂的固态与溶液态分为两种测试方式:①将少量待测粉末研磨后与KBr 混合,压成薄片后进行测试;②将凝胶剂的氯仿溶液涂在KBr 压片上直接进行测试.X 射线衍射(XRD)图谱由布鲁克D8-S4 射线衍射仪测试得到,扫描速度0.2 s/步,2θ=2°~35°,步长0.02°.凝胶剂分子的最优化结构采用Gaussian 软件获得,计算方法为密度泛函法(DFT).扫描电子显微镜结果使用日立S-4800 场发射扫描电子显微镜观察得到.流变学测试采用Anton Paar Physica MCR 301 流变仪,25 ℃下将室温凝胶样品置于流变仪与平行板之间进行测试.
1.2 凝胶剂的制备
1.2.1 2,4-(3,4-二氯苯亚甲基)-D-葡萄糖酸酰十六胺(凝胶剂G16)的合成
在500 mL 烧瓶中分别加入10 g(0.028 mol)3,4-二氯苯亚甲基-D-葡萄糖酸甲酯和80 mL 甲醇,搅拌20 min 后加入 20.28 g(0.084 mol)十六胺和 0.02 g DMAP.室温下剧烈搅拌 12 h,随后加入 50 mL水.搅拌30 min 后进行抽滤,滤饼用水洗涤两次得到粗品.65 ℃下将粗品在甲醇中重结晶,烘干得到产物,产率为60%.图1 为凝胶剂G16 的分子结构.
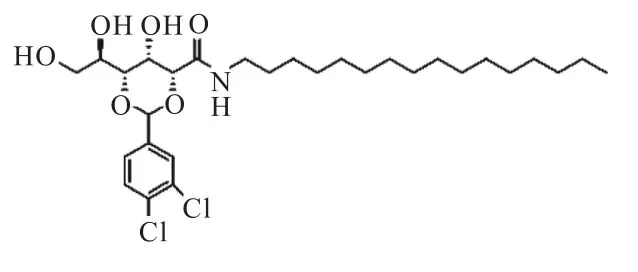
图1 凝胶剂G16的分子结构Fig.1 Gelator structure of G16
1.2.2 2,4-(3,4-二氯苯亚甲基)-D-葡萄糖酸酰十六胺-20℃-甲醇(G16-20-Me)等凝胶剂的制备
在装有磁子的200 mL 烧杯中,向100 mL 甲醇溶剂中加入0.5 g 的2,4-(3,4-二氯苯亚甲基)-D-葡萄糖酸酰十六胺(凝胶剂G16).搅拌加热至50 ℃下保持30 min,以使G16 粉末完全溶解于甲醇中.待完全溶解后将溶液冷却至 20 ℃形成饱和甲醇溶液.20 ℃下放置待甲醇完全挥发,样品从溶剂中析出.得到凝胶剂2,4-(3,4-二氯苯亚甲基)-D-葡萄糖酸酰十六胺-20 ℃-甲醇(简称为G16-20-Me).
保持所用溶剂甲醇不变,将组装温度由20 ℃分别调整为30 ℃和40 ℃,得到凝胶剂分别称为G16-30-Me 与G16-40-Me.保持温度20 ℃不变,将所用溶剂分别替换为乙醇和四氢呋喃,得到凝胶剂分别称为G16-20-Et 与G16-20-THF.
1.3 凝胶性能测试
1.3.1 室温凝胶性能
首先将一定量的凝胶剂加入试管中,再添加定量的溶剂.在室温下将试管静置8 h,将试管翻转观察试管内的“溶液”是否仍能流动.当不存在重力流动时,则判定发生了室温凝胶化.
1.3.2 粉末最低凝胶浓度(PCGC)测定
首先将一定量的凝胶剂加入试管中,再添加定量的溶剂.在室温下将试管静置8 h,若形成凝胶,则将溶剂的量增加0.1~0.2 mL 重新进行测定.直到不能形成凝胶时,上一次的凝胶剂浓度则称之为粉末最低凝胶浓度(PCGC).
2 结果与讨论
2.1 室温凝胶性能
几种凝胶剂的室温凝胶性能测试结果如表1 所示.65 ℃下从甲醇中自组装得到的凝胶剂G16,不具备室温凝胶性能.而20 ℃下从甲醇中自组装得到的凝胶剂G16-20-Me 在氯苯、甲苯等芳烃溶剂以及环己烷中具有优异的室温凝胶性能,粉末最低凝胶浓度在18~35 mg/mL 之间.保持自组装所用溶剂甲醇不变,将自组装的温度调整至30 ℃与40 ℃,发现30 ℃下形成的凝胶剂G16-30-Me 仅可凝胶环己烷一种溶剂,40 ℃下形成的凝胶剂G16-40-Me 不具备室温凝胶性能.这表明,较低的自组装温度有利于形成室温凝胶剂.保持自组装温度20 ℃不变,将自组装所用溶剂甲醇替换为乙醇和四氢呋喃.发现乙醇溶剂下形成的凝胶剂G16-20-Et 对芳烃及环己烷同样具有室温凝胶性能,粉末最低凝胶浓度也与G16-20-Me接近.而四氢呋喃溶剂下形成的凝胶剂G16-20-THF不具备室温凝胶性能.这表明,自组装所用溶剂会影响室温凝胶剂性能.保持20 ℃、甲醇溶剂的自组装条件不变,将凝胶剂G16 替换为烷基链较短的凝胶剂2,4-(3,4-二氯苯亚甲基)-D-葡萄糖酸酰辛胺,则形成的凝胶剂不具备室温凝胶性能,这表明烷基链的长短会影响室温凝胶性能.而各凝胶剂在水中都不具备凝胶能力,这为凝胶剂在废水中对有机溶剂的室温相选择凝胶化的实际应用奠定了基础.
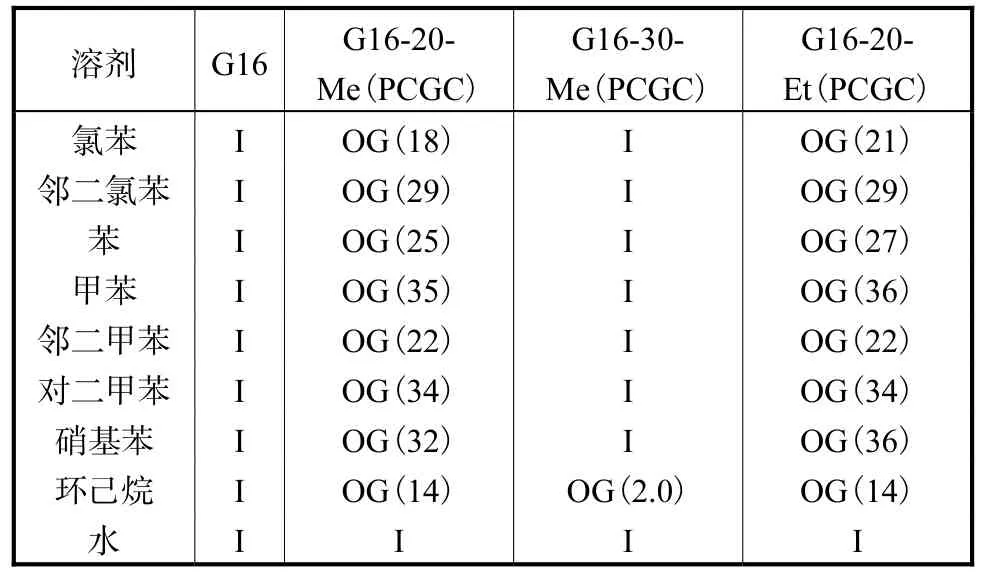
表1 各凝胶剂在不同溶剂中的室温凝胶性能Tab.1 Room-temperature gelation properties of various gelators in different solvents

图2 G16-20-Me 氯仿溶液、氯苯干凝胶及各凝胶剂粉末的红外光谱图Fig.2 FT-IR spectrograms of G16-20-Me chloroform solution,chlorobenzene xerogel,and various gelator powders
2.2 傅里叶红外光谱(FT-IR)
为研究凝胶剂自组装过程中的主要驱动力,对凝胶剂G16-20-Me 进行了游离态和干凝胶态(质量浓度为20 mg/mL)的红外光谱测试.从图2(a)可以看出,氯仿溶液中的3 452 cm-1、1 647 cm-1、2 928 cm-1和2 860 cm-1分别为OH(NH)的吸收峰、C=O 的吸收峰、CH2的不对称伸缩振动(νas)和对称伸缩振动(νs)的吸收峰,而在氯苯干凝胶中,分别转移到了3 396 cm-1、1 635 cm-1、2 923 cm-1和2 852 cm-1.这些变化说明OH(NH)和C=O 形成了分子间氢键参与了自组装,并且烷基链之间存在着范德华作用力.此外,由前期工作可知,π-π 堆积作用也存在于凝胶剂自组装过程中[4].
为了解室温凝胶体系的机理,对各凝胶剂粉末进行了进一步的研究.从图2(b)中可以看出,G16、G16-20-THF、G16-40-Me 粉末的CH2的不对称伸缩振动(νas)以及对称伸缩振动(νs)的吸收峰在2 921 cm-1和2 850 cm-1处,而在G16-20-Me、G16-20-Et、G16-30-Me 粉末中则移动到了 2 923 cm-1和2 852 cm-1处.此外,G16-20-Me 粉末与G16-20-Me氯苯干凝胶的吸收峰均出现在相同的位置.这说明G16、G16-20-THF、G16-40-Me 粉末中烷基链之间的范德华作用力较强,而G16-20-Me、G16-20-Et、G16-30-Me 粉末中烷基链之间的范德华作用力较弱.
2.3 X射线衍射(XRD)
为了进一步探究凝胶剂分子的堆积结构,对G16-20-Me 的氯苯干凝胶(质量浓度为20 mg/mL)进行了X 射线衍射实验,如图3(a)所示,G16-20-Me 氯苯干凝胶图像在2.91 nm、1.45 nm 和0.97 nm 处出现3 个峰,接近1∶1/2∶1/3 的比例,表明分子为层状堆积.根据布拉格方程,层间距为2.91 nm.
如图3(b)所示,各凝胶剂粉末的XRD 测试结果显示,G16-20-Me 粉末与G16-20-Me 氯苯干凝胶的XRD 图谱基本一致,都为层状堆积,且分子层间距都为2.91 nm.此外,由DFT 计算得到G16 分子结构的最优化构型长度为2.52 nm.凝胶剂G16-20-Me 的分子层间距介于单分子长度及双倍分子长度之间,这表明烷基链之间发生了相互交叉.而G16 粉末的分子层间距为2.83 nm,小于G16-20-Me 粉末的层间距,这说明G16-20-Me 粉末的烷基链交叉程度更低,如图4 所示.G16-20-Et、G16-30-Me 粉末的分子层间距与G16-20-Me 粉末接近,分别为2.91 nm 和2.87 nm,而G16-20-THF、G16-40-Me 粉末的分子层间距与G16 粉末相同,都为 2.82 nm.此外,G16-20-Me、G16-20-Et 粉末在XRD 图谱广角区域的衍射峰较宽,G16-30-Me 次之,说明G16-20-Me、G16-20-Et、G16-30-Me 粉末的结晶性较低.而 G16、G16-20-THF、G16-40-Me 粉末在XRD 图谱广角区域的峰较为尖锐,说明G16、G16-20-THF、G16-40-Me 粉末的结晶性更高,分子排列较为规整.
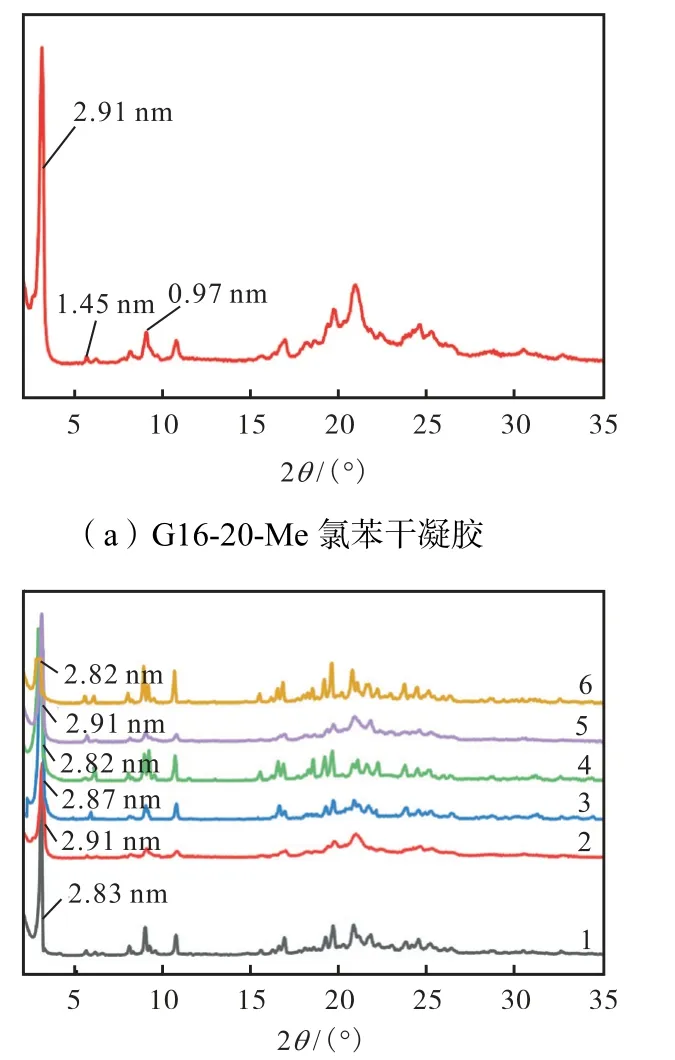
图3 G16-20-Me 氯苯干凝胶及各凝胶剂粉末的XRD 光谱图Fig.3 XRD patterns of G16-20-Me chlorobenzene xerogel,and various gelator powders
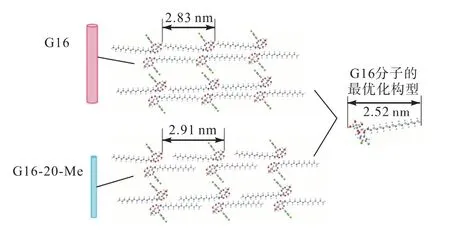
图4 G16 分子的最优化构型及凝胶剂G16、G16-20-Me的分子堆积结构Fig.4 Optimum configuration of G16 molecule and molecular packing structures of gelators G16 and G16-20-Me
2.4 场发射扫描电镜(SEM)
为研究各凝胶剂粉末及G16-20-Me 氯苯干凝胶(质量浓度为20 mg/mL)的微观形貌,进行了SEM 测试.从图5 可以看出,凝胶剂G16-20-Me 和G16-20-Et 粉末均为较细的针状纤维,平均直径约为0.07 μm,平均长度约为1.12 μm,纤维间孔隙较大,具有疏松的三维网状结构.凝胶剂G16、G16-40-Me 和G16-20-THF 粉末均为较粗的棒状纤维,平均直径约为0.84 μm,平均长度约为10.78 μm,纤维间几乎无孔隙,为一维纤维束紧密堆积结构.凝胶剂G16-30-Me 的纤维大小介于上述两种结构之间.由结晶性较低导致的纤维细小及结构疏松使得凝胶剂与溶剂分子接触更加充分,更容易被溶剂分子破坏,从而具有室温凝胶性能.值得注意的是,G16-20-Me 氯苯干凝胶的纤维直径与G16-20-Me 粉末基本相同.结合G16-20-Me 氯苯干凝胶与G16-20-Me 粉末的红外光谱图及XRD 图谱同样保持一致,这里可以推测出:在室温凝胶过程中,凝胶剂粉末纤维与溶剂的接触使纤维间的连接点断裂,而粉末纤维的内部组装结构没有变化.
基于以上分析,得出了室温凝胶自组装机理.如图6 所示,在氢键和π-π 堆积的作用下,凝胶剂分子交替排列形成一维组装体,一维组装体通过范德华作用力进一步形成纤维.对于凝胶剂G16 而言,由于是在65 ℃下甲醇中自组装形成的,结晶性较高,纤维较大且堆积紧密,组装体间的范德华作用力较强.因此,需要通过加热-冷却过程将强作用力破坏然后重新进行组装.对于凝胶剂G16-20-Me 而言,组装温度为20 ℃,较低的温度使其结晶性较低,纤维细小且结构疏松,组装体间的范德华作用力较弱.室温下,纤维连接点处的弱范德华作用力很容易被溶剂破坏,当达到一定浓度时,分散的纤维在纤维表面的范德华作用力下又自发地交织在一起,最终得到优异的室温凝胶性能.而凝胶剂2,4-(3,4-二氯苯亚甲基)-D-葡萄糖酸酰辛胺因其烷基链较短,采用相同自组装条件形成的凝胶剂结晶性较强,组装体难以被溶剂分子破坏,因此不具有室温凝胶性能.

图5 各凝胶剂粉末及G16-20-Me氯苯干凝胶的SEM图像Fig.5 SEM images of various gelator powders and G16-20-Me chlorobenzene xerogel
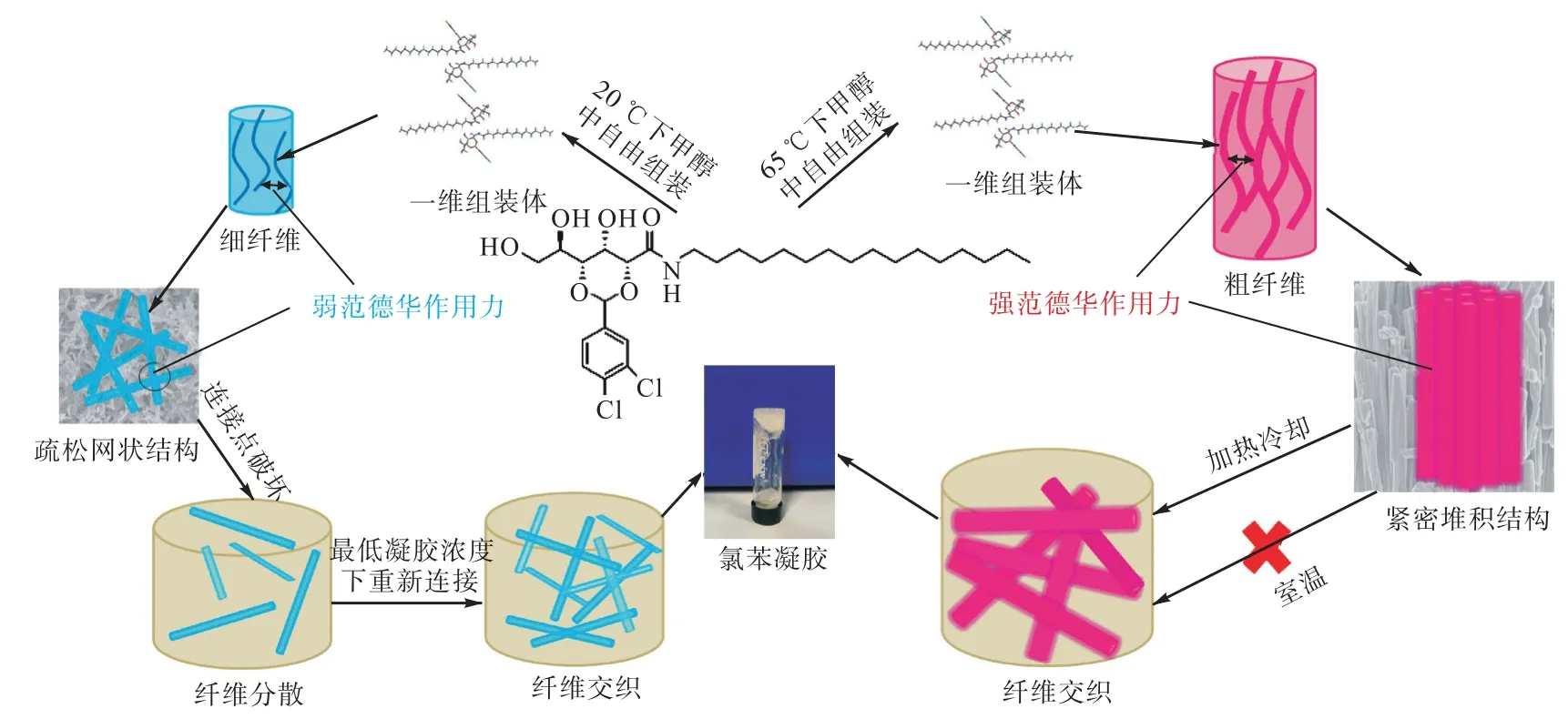
图6 凝胶剂G16、G16-20-Me的凝胶模型Fig.6 Gel model of gelators G16 and G16-20-Me
2.5 流变性能
在凝胶测试过程中发现,所有的G16-20-Me 室温凝胶在受到机械破坏后,可在10 s 内迅速自我修复,如图7(a)所示,将G16-20-Me 的氯苯凝胶(质量浓度为20 mg/mL)大力摇动直到具有流动性,然后在室温下静置1 min,凝胶重新形成.此外,G16-20-Me的氯苯凝胶还具有良好的黏弹性,如图7(b)所示,从注射器挤出.
为进一步研究G16-20-Me 凝胶在室温下的力学性能,进行了流变学测试.如图8(a)、(b)所示,G16-20-Me 的氯苯凝胶及甲苯凝胶(质量浓度均为40 mg/mL)在应变扫描的线性黏弹区域(LVR)中,储能模量(G′)均大于损耗模量(G″),表明G16-20-Me的氯苯凝胶及甲苯凝胶均为真凝胶,且具有一定的稳定性.G16-20-Me 氯苯凝胶的流动点为45%,大于G16-20-Me 甲苯凝胶的流动点25%,表明G16-20-Me 氯苯凝胶的黏弹性更好.如图8(c)、(d)所示,首先将凝胶置于0.1%的应变下,储能模量(G′)大于损耗模量(G″),表现为固体性质.然后对凝胶施加100%的应变以破坏凝胶,一段时间后再对凝胶施加0.1%的应变,重复2 个周期.储能模量(G′)及损耗模量(G″)在破坏停止后可立即恢复至原来的值,这说明G16-20-Me 凝胶具有良好的自修复性.

图7 G16-20-Me凝胶的自修复性及黏弹性Fig.7 Self-healing and viscoelastic properties of G16-20-Me chlorobenzene gel

图8 G16-20-Me氯苯凝胶和甲苯凝胶的流变学测试结果Fig.8 Rheological tests of G16-20-Me chlorobenzene gel and toluene gel

图9 G16-20-Me粉末对氯苯的回收过程Fig.9 Recovery of G16-20-Me powder for chlorobenzene
2.6 室温相选择凝胶法去除有机污染物
使用凝胶剂G16-20-Me 进行了水中有机污染物的去除及回收实验,如图9 所示,在含有3 mL 水的玻璃小瓶中加入3 mL 氯苯,由于氯苯的密度大于水,氯苯层沉入水层下方.将60 mg G16-20-Me 粉末直接添加到氯苯/水的混合物中并进行简单的摇晃,G16-20-Me 粉末即可进入氯苯层.在室温下静置10 min,形成氯苯凝胶.凝胶块很容易用镊子夹出来,并通过简单地蒸馏将氯苯进行回收,凝胶剂经重结晶后可重新利用.这是首例在回收氯苯中具有实际应用潜力的凝胶剂.经测试,其他芳烃溶剂均可通过此种方式进行回收.
3 结 语
通过对2,4-(3,4-二氯苯亚甲基)-D-葡萄糖酸酰十六胺(凝胶剂G16)的自组装条件进行调控,使其在20 ℃甲醇中进行自组装形成粉末型相选择凝胶剂G16-20-Me,该粉末凝胶剂对芳烃等溶剂具有出色的室温凝胶能力.通过红外光谱、X 射线衍射实验、扫描电子显微镜及理论计算表明,分子间的自组装驱动力为氢键、π-π 堆积和范德华作用力.对凝胶剂自组装条件的调控降低了凝胶剂的结晶性,形成细小的纤维及疏松的结构,组装体间的范德华作用力减弱.室温下,纤维连接点处的弱范德华作用力很容易被溶剂破坏,当达到一定浓度时,分散的纤维在纤维表面的范德华作用力下又自发地交织在一起,最终得到优异的室温凝胶性能.此外,流变学实验表明G16-20-Me凝胶具有优异的自修复性及黏弹性.对G16-20-Me粉末进行了氯苯的回收实验,证明该凝胶剂在含芳烃废水处理领域具有潜在的应用价值.
