高邻位酚醛基纳米活性碳纤维制备及其吸附性能
2021-01-05张啸梅焦明立贾万顺张彩云
杨 凯, 张啸梅, 焦明立, 贾万顺, 刁 泉, 李 咏, 张彩云, 曹 健
(1. 中原工学院 服装学院, 河南 郑州 450007; 2. 中原工学院 材料与化工学院, 河南 郑州 450007)
酚醛纤维是20世纪70年代出现的第1种交联型合成纤维[1],独特的三维网状结构赋予其阻燃、耐烧蚀、耐辐照等特性[2]。酚醛纤维炭化可得到无定型碳,具有孔隙率高而密度低的特点,成为制备多孔活性碳材料的理想前驱体[3-4]。交联的酚醛纤维不需预氧化步骤,可直接炭化,炭化过程具有效率高、污染小、残碳率高等特点[5],所制备的酚醛基活性碳纤维具有吸附容量大、吸/脱附速度快、强度高、断裂伸长率大等优点,可应用于超级电容器电极、气/液吸附材料、储氢及药物载体等领域[6-7]。
随着静电纺丝技术的快速发展,利用溶液/熔体静电纺丝技术制备热固性或热塑性酚醛及其共混物纳米纤维得到了广泛的研究,并实现了快速制备形貌可控的纳米级酚醛纤维,所得纤维表现出良好的炭化和活化性能[8-10],但对静电纺高邻位酚醛的研究较少。为此,本文通过合成结构规整、黏度小、易纺丝的高邻位热塑性酚醛树脂,并通过静电纺丝、溶液固化、炭化及活化步骤,得到高邻位酚醛基纳米活性碳纤维,研究了酚醛纤维在各加工过程中结构的变化,并比较了碳纤维、活性碳纤维孔结构及吸附性能的变化。
1 实验部分
1.1 实验材料
甲醛水溶液,甲醛质量分数为37%~40%,西陇化工股份有限公司;聚乙烯醇缩丁醛(PVB),分析纯,阿拉丁试剂上海有限公司;苯酚、氢氧化钠,分析纯,天津市恒兴化学试剂制造有限公司;乙酸锌,分析纯,天津市科密欧化学试剂有限公司;浓硫酸,衡阳市凯信化工试剂有限公司;无水乙醇、四氢呋喃,分析纯,天津市富宇精细化工有限公司;盐酸(HCl)水溶液,HCl质量分数为36%~38%,洛阳市昊华化学试剂有限公司;氢氧化钾(KOH),分析纯,天津市风船化学试剂科技有限公司。
1.2 实验方法
1.2.1 高邻位热塑性酚醛树脂的制备
称取苯酚、甲醛及乙酸锌(三者质量分别为4、200、146.6 g)加入三口烧瓶中,沸腾反应4 h,滴加1.1 g浓硫酸后保持1 h,真空脱水得到高邻位热塑性酚醛树脂(PR)[11]。
1.2.2 静电纺酚醛基纳米活性碳纤维的制备
称取一定配比的PR及PVB溶于乙醇和四氢呋喃混合溶剂(乙醇和四氢呋喃体积比为4∶1)中,为探讨合适的静电纺丝液黏度,按表1所示配比配制PR和PVB溶液,待溶液混合均匀后,采用TL-01型静电纺丝机(深圳通力微纳科技有限公司)进行静电纺丝,得到酚醛初生纤维(PF-As),其中纺丝电压为19 kV,接收距离为18 cm,进给速率为1.0 mL/h。
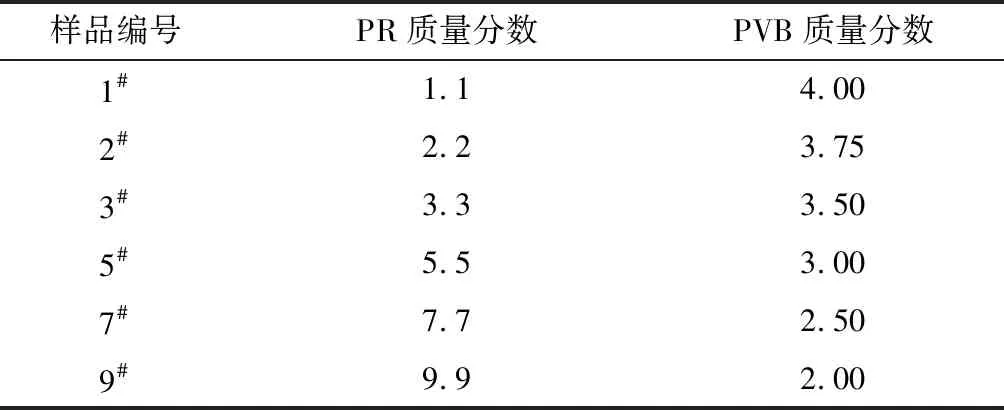
表1 纺丝原液中PR和PVB的配比Tab.1 Content of PR and PVB in spinning dope %
将酚醛初生纤维置于固化溶液(盐酸质量分数为18.5%、甲醛质量分数为12%的水溶液)中,以0.5 ℃/min 升温至95 ℃后保持2 h,自然降温,水洗干燥后得到高邻位酚醛纤维(PF-S);然后,将高邻位酚醛纤维在N2氛围中以5 ℃/min升温至900 ℃ 炭化1 h, 制得酚醛基碳纤维(PCF),其中在升温至240 ℃时采样进行相应测试(该阶段样品记为PF-240 ℃); 最后,浸泡于KOH溶液中10 h(碱碳比为4∶1),取出干燥并在N2中以5 ℃/min升至800 ℃活化1 h,酸洗后水洗至中性,干燥得到高邻位酚醛基纳米活性碳纤维(PACF)。
后文样品编号采用不同制备阶段试样名称和表1 中样品编号表示,如PF-As-1#即表示按照表1中1#样品配比制备的酚醛初生纤维,依此类推。
1.3 测试方法
1.3.1 化学结构测试
采用溴化钾压片法,使用Nicolet iS50 FT-IR型红外光谱分析仪(FT-IR,美国赛默飞公司)测试样品的化学结构。测试波数范围为4 000~400 cm-1,扫描次数为32。
1.3.2 表面形貌观察
使用JSM-6360LV型扫描电子显微镜(SEM,日本电子株式会社)观察喷金处理后的试样形貌,测试电压为25 kV,电流为10 mA。
1.3.3 热稳定性能测试
使用STA449-F5型热重同步分析仪(TG,德国耐驰公司),在N2气氛中以10 ℃/min的升温速率从20 ℃ 升温至900 ℃进行热稳定性能分析。
1.3.4 比表面积及孔径测试
使用JW-BK100B型比表面积及孔径分析仪(北京精微高博科学技术有限公司),测试试样在77 K 下的N2吸附/脱附等温线,测试前纯化气路并脱气除水。
1.3.5 吸附性能测试
标准曲线的计算:配制亚甲基蓝(MB)和碘标准溶液,利用UV-2600型紫外分析仪(日本岛津公司)测试不同浓度标准溶液的吸光度(MB对应波长为665 nm,碘对应波长为350 nm),并拟合出MB和碘溶液的吸光度与浓度的关系,分别为:
AMB=0.062 5CMB
(1)
AI=5 949.956 6CI-0.888 1
(2)
式中:AMB、AI分别为MB和碘的吸光度;CMB为MB的质量浓度,mg/L;CI为碘的浓度,mol/L。
MB平衡吸附值[12]的测定:将适量试样放入装有不同体积的100 mg/L亚甲基蓝溶液的碘量瓶中,在30 ℃恒温水浴中搅拌5 h后过滤;然后测定滤液的吸光度,并由式(1)计算亚甲基蓝的平衡质量浓度,根据下式计算亚甲基蓝吸附量:
QMB=(V0-CMB)V/m
(3)
式中:QMB为亚甲基蓝吸附量,mg/g;V0为亚甲基蓝溶液的初始质量浓度,其值为100 mg/L;V为亚甲基蓝溶液体积,mL;m为试样的质量,mg。
碘吸附值[13]测定:取适量配制好的0.1 moL/L的碘标准溶液稀释100倍,加入试样充分搅拌50 min 使其达到吸附平衡,过滤后用紫外分析仪测定吸光度,根据下式计算碘吸附值:
QI=(C0-CI)VWI/m
(4)
式中:QI为碘吸附量,mg/g;C0为碘溶液的初始浓度,moL/L;V为碘溶液的体积,mL;m为试样的质量,g;WI为碘的相对分子质量,其值为253.809。
2 结果与讨论
2.1 化学结构分析
图1示出不同制备阶段酚醛试样的红外光谱图。由PVB曲线可知, 3 454 cm-1处对应未反应的羟基的伸缩振动峰,2 957和2 868 cm-1处分别对应CH2和CH中C—H伸缩振动峰,1 124 cm-1处为C—O的伸缩振动峰,995 cm-1处也出现了PVB的特征吸收峰。PR曲线中3 389、1 600 cm-1处分别对应酚羟基、苯环的伸缩振动峰,1 457、1 239 cm-1处分别对应亚甲基键桥的弯曲振动及醚键的伸缩振动峰。

图1 不同制备阶段酚醛试样的红外光谱图Fig.1 FT-IR spectra of phenolic samples at different preparation stage
酚醛初生纤维(PF-As-3#)曲线中分别出现了PVB和PR的吸收峰,但溶液固化后PVB的特征吸收峰降低或消失(见图中 PF-S-3#曲线),包括2 957 cm-1处亚甲基峰,1 124 cm-1处的C—O伸缩振动峰,以及995 cm-1处的PVB特征吸收峰都出现了下降。这是由于PVB在酸性溶液中发生醇解反应生成了聚乙烯醇链段,从而在1 648 cm-1处出现强烈的—OH弯曲振动峰;同时固化液中盐酸、甲醛的存在又为缩甲醛反应提供了条件,如图1中PF-S-3#曲线在1 175、1 076、1 020 cm-1处出现了吸收峰的增加,其中1 175、1 076 cm-1处分别对应缩甲醛的特征吸收峰,1 020 cm-1处对应聚乙烯醇缩甲醛的C—O伸缩振动峰,从而可推测在酸性甲醛溶液固化后,也出现了部分PVB转变为聚乙烯醇缩甲醛链段的现象。
在红外光谱图中,832、752 cm-1处的吸收峰分别对应酚环中邻位、对位取代,其吸收峰强度的变化反映了苯环中邻位取代和对位取代比例的改变[14-15]。本文合成的PR在752 cm-1处具有明显的邻位取代吸收峰,证明合成的是高邻位酚醛树脂。通过对比纺丝、溶液固化和240 ℃热处理的酚醛纤维红外光谱(见图1中 PF-As-3#、PF-S-3#、PF-240 ℃-3#)可知,随着实验过程的进行,752 cm-1处吸收峰的强度不断降低。说明随着纺丝、固化过程的进行,更多的对位活性点参与反应,使得酚醛中邻位取代比例下降,同时对位取代反应程度的增加也使得酚环的三官能团得到充分利用,形成了交联度逐渐增加的酚醛纤维。
由图1 中PCF-3#、PACF-3#的红外图谱可知:炭化和活化后,酚醛试样大部分的吸收峰消失,说明通过炭化和活化后纤维中极性基团消失,形成了大量C—C 非极性键连接的炭材料;在3 447、1 574和1 132 cm-1处出现的较弱吸收峰则是由于PCF-3#、PACF-3#中残留部分的羧酸基团及表面吸附气体产生的。
2.2 表面形貌分析
图2为不同PR和PVB配比的PACF纤维的表面扫描电镜照片。可知:当PR质量分数较低时,纤维出现溶并现象;随着纺丝液中PR质量分数的增加,溶并现象消失,PACF纤维直径不断增大,但直径均匀性呈先增加后降低趋势;同时随着PR质量分数的增加,PACF纤维出现断裂点,纤维韧性降低。由于初生纤维制备过程中,乙醇和四氢呋喃作为混合溶剂挥发快,易形成皮芯结构,使最终得到的纤维呈扁平带状[16],其中PACF-3#中带状纤维厚度约为250 nm。

图2 不同PR和PVB配比的PACF纤维的扫描电镜照片Fig.2 SEM images of PACF fiber with different PR and PVB content
2.3 热性能分析
图3示出不同制备阶段酚醛试样的TG曲线。由 PR的TG曲线可知:在100~340 ℃范围内,由于酚醛树脂中未反应小分子的逸出和弱末端基团的脱除引起了低温质量损失;而340~470 ℃范围的质量损失是由树脂中酚羟基基团间缩聚生成二苯醚的脱水反应,及酚羟基基团与亚甲基键桥间缩合生成次甲基交联点的脱水反应所致;高于470 ℃时,PR的质量损失来源于亚甲基键桥氧化形成羰基基团,苯环间缩聚形成多环芳烃的反应等[17]。
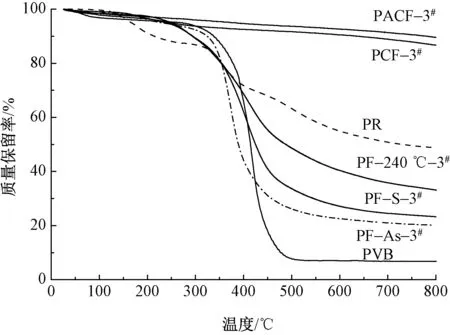
图3 不同制备阶段酚醛试样的TG曲线Fig.3 TG curves of phenolic samples at different preparation stage
由图3中 PF-As-3#、PF-S-3#及PF-240 ℃-3#的TG曲线可知:在240 ℃以下的极小质量损失主要是未反应小分子的挥发引起的;240 ℃以上是主要的质量损失阶段,包括酚醛的分子间脱水及环化脱氢、炭化过程,PVB及其解聚产物的分解。其中经过溶液固化及240 ℃处理的PF-240 ℃-3#纤维在240 ℃以上首先出现了质量损失,这可能是由于PVB转化产物聚乙烯醇嵌段首先分解导致的。在800 ℃时,与PF-As-3#初生纤维相比,PF-S-3#、PF-240 ℃-3#的质量损失依次减少,240 ℃处理酚醛纤维的质量损失明显降低,残碳率达到33%。说明炭化过程的低温段(小于240 ℃)进一步提高了纤维交联度,有利于残碳率的提升;同时也说明本文合成的高邻位酚醛树脂,其酚环对位的高反应性有利于进行交联反应,生成交联结构均匀的酚醛纤维,有利于制备高残碳率的PCF及PACF,该交联结构也保证了无需热氧化即可进行炭化处理过程。由于PF-As-3#、PF-S-3#及PF-240 ℃-3#纤维中含有的PVB在加热过程中完全分解,因此,其质量损失率均比酚醛树脂的高。PCF-3#和PACF-3#由于在制备过程中已在900 ℃ 下N2氛围中进行炭化处理,因此,在TG温度范围内未见明显质量损失。
2.4 比表面积及孔径分析
图4示出不同PR和PVB配比制备的PCF及PACF在77 K下的氮气吸附/脱附等温线及孔径分布图。由图4(a)、(b)可知,不同配比的PCF及PACF等温线均为典型的I型等温线,表明所得PCF及PACF均为典型的微孔型材料[18-19]。由图4(a)可知:PCF纤维的吸附主要发生在相对压力(P/P0, 其中P为平衡压力(kPa),P0为饱和蒸汽压(kPa))小于0.1的阶段(低压段),其吸附量会随着P/P0的增大而快速增加;在0.1
表2示出不同PR和PVB配比的PCF及PACF的比表面积及孔结构参数。由图4(a)和表2可知,随着PVB质量分数的降低,PCF纤维氮气吸附量呈先上升后下降的趋势,其中PCF-3#纤维的氮气吸附量达到最大值。这是由于高邻位酚醛基碳纤维在900 ℃ 炭化过程中,PVB分解成小分子逸出产生孔洞:当PVB质量分数过大时,其造孔能力过强,纤维中孔间贯穿造成孔坍塌,导致纤维比表面积和孔体积下降,从而使氮气吸附量下降;PVB质量分数较小时,纤维形成的孔洞较少,比表面积和孔体积减少,相应的氮气吸附量亦会降低。
注:图(c)和(d)纵坐标中V为孔容,cm3/g;D为平均孔径,nm。图4 不同PR和PVB配比的PCF及 PACF纤维的氮气吸附/脱附等温线和孔径分布Fig.4 N2 adsorption/desorption isotherms and pore size distribution of PCF and PACF fiber with different PR and PVB content.(a)N2 adsorption/desorption isotherms of PCF;(b)N2 adsorption/desorption isotherms of PACF;(c)Pore size distribution of PCF;(d)Pore size distribution of PACF

表2 不同PR和PVB配比的PCF 和 PACF纤维的孔结构参数Tab.2 Textural properties of PCF and PACF fiber with different PR and PVB content
由图4(b)氮气吸附/脱附等温线可知:PACF由于经过了活化处理,其介孔量多于PCF,且有较宽的介孔孔径分布,因而PACF均出现明显的滞后圈;此外,PACF在相对压力为0.1附近的等温线斜坡较为平缓。说明活化过程中,由于与微晶碳网平面平行方向的边缘面易受KOH攻击,导致微晶不均匀气化及其外表面碳元素的脱离,从而使碳纤维的烧蚀程度加重,形成了新的孔洞,最终PACF比表面积增大[20]。由图4(a)、(b)对比可知,碳纤维经活化后获得了丰富的微孔和较大的孔径分布及孔容,使得PACF的氮气吸附量远远高于PCF。
PCF和PACF的孔径分布图(见图4(c)、(d))也进一步证实了以上结论。从图4(c)可知,不同PR和PVB配比的PCF在0.6~10 nm范围内均匀分布着一定的微孔及介孔。其中:在0.59~0.61 nm 范围内分布着大量的超微孔(<0.7 nm); 在0.7~1.38 nm 范围内随着孔径的增大,微孔含量不断下降;而在1.38~1.91 nm范围内的微孔含量又逐渐升高,并在1.91 nm处达到极值;PCF-2#及PCF-3#含有的介孔较多。由图4(d)可知,随着PACF烧蚀程度的增加,超微孔含量最低点移至1.58 nm处,含量最高点由1.88 nm 升至2.05 nm处,大量微孔、介孔的孔径增大,使PACF的比表面积和孔容增加。
从表2也可以看出:PCF的比表面积和孔容随着PVB质量分数的减小,均呈现先增加后降低趋势,说明介孔含量及平均孔径逐渐减少;当PVB质量分数为3.5%(PCF-3#)时,纤维微孔丰富,比表面积为712 m2/g,孔容为0.344 cm3/g。而经过活化后PACF的比表面积、孔容、微孔比表面积及孔容均大幅高于PCF;且随着PVB质量分数的减小,PACF的比表面积和孔容也出现先增大后减小的趋势,同样在PACF-3#样品配比时达到最佳,该条件下其比表面积为1 409 m2/g,孔容为0.771 cm3/g。
2.5 亚甲基蓝及碘吸附量分析
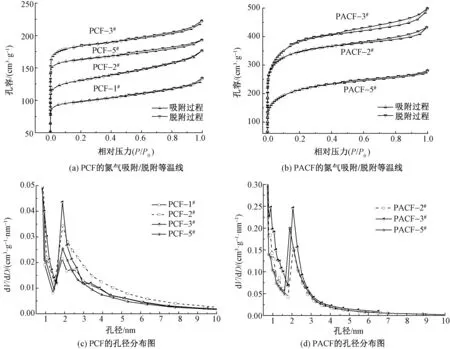
使用亚甲基蓝和碘进行吸附性能测试,进一步研究酚醛基碳材料对小分子的吸附性能。亚甲基蓝分子直径一般为碘分子的3倍左右,其吸附值常用来表征吸附剂表面1.5~2.8 nm的孔径,其中起主要作用的是1.6 nm附近的孔径[21]。碘分子直径在0.49~0.54 nm范围,碘吸附主要发生在大于1 nm的孔表面上,其中起主要作用的是1.1 nm附近的孔径,二者常用来表征多孔碳材料对非极性有机小分子的吸附能力。不同PR和PVB配比的PCF及PACF纤维对亚甲基蓝及碘的吸附量的变化如图5所示。
从图5(a)可知,随着PVB质量分数的降低,PCF对亚甲基蓝及碘的吸附量整体呈先增大后降低趋势,并在PCF-3#配比(PVB质量分数为3.5%)时达到最大值,这与2.4节得到的PCF-3#纤维在0.7~2.0 nm存在较多微孔的结论相符。该配比下,PCF的亚甲基蓝吸附量为524 mg/g,碘吸附量为1 970 mg/g。
由图5(b)可知,不同PR和PVB配比的PACF纤维对亚甲基蓝及碘吸附量均高于PCF,但吸附趋势一致,即随着PVB质量分数的减少,亚甲基蓝及碘吸附量呈先增加后降低趋势,同样在PACF-3#配比时达到对亚甲基蓝及碘的吸附量极值,此时亚甲基蓝吸附量为837 mg/g,碘吸附量为2 641 mg/g,表现出对小分子优异的吸附性能。

图5 不同PR和PVB配比的PCF和PACF纤维的碘及亚甲基蓝吸附量Fig.5 Iodine and MB adsorption capacity of PCF and PACF fiber with different PR and PVB content
2.6 亚甲基蓝及碘吸附量与吸附时间关系
研究PACF-3#样品吸附过程中吸附时间对亚甲基蓝及碘吸附量的影响,结果如图6所示。可知,PACF-3#对亚甲基蓝及碘的吸附过程均符合多孔吸附剂的液相吸附过程。图6(a)曲线可分为3个阶段:0~0.5 h阶段(液膜扩散段)为亚甲基蓝分子通过溶液扩散快速吸附到纤维表面;0.5~3 h阶段(颗粒扩散段)为亚甲基蓝分子逐步向内部孔隙扩散,此阶段吸附速率下降;最后为亚甲基蓝分子吸附在纤维孔内吸附位上,吸附趋于平衡,亚甲基蓝吸附量为837 mg/g, 达到吸附平衡。图6(b)碘吸附过程也可分为0~5 min的快速吸附阶段,5~40 min 的逐步吸附阶段及40~50 min吸附平衡阶段,并在40 min 达到吸附平衡,碘吸附量为2 641 mg/g。 说明PACF对碘具有较好的吸附速率和吸附容量。
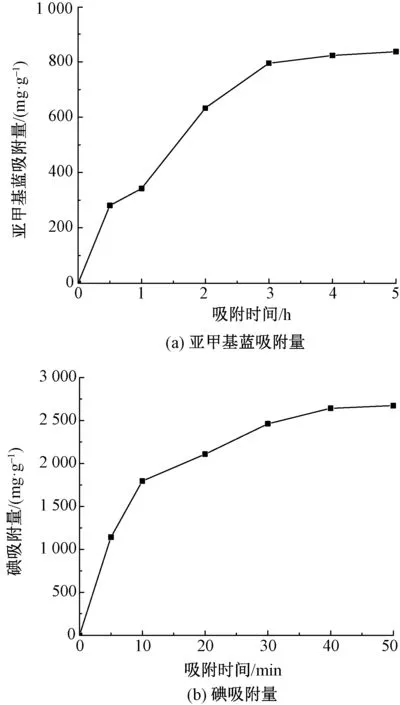
图6 吸附时间对PACF-3#纤维亚甲基蓝、碘吸附量的影响Fig.6 Effect of adsorption time on MB (a) and iodine (b) adsorption capacity of PACF-3# fiber
3 结 论
本文通过合成高邻位酚醛树脂为前驱体,与聚乙烯醇缩丁醛(PVB)共混后进行溶液静电纺丝,制备得到酚醛初生纤维,然后经溶液固化、炭化、活化后得到了高邻位酚醛基纳米活性碳纤维。研究发现:初生纤维经过固化交联后,PVB发生了部分醇解,使酚醛纤维的低温热稳定性升高;同时由于交联反应在纤维内形成了网络结构,使得酚醛纤维高温热稳定性也有提高,可不经预氧化而实现一步炭化处理,并经活化后制备得到扁平状纳米活性碳纤维。不同酚醛基碳纤维及活性碳纤维的比表面积、孔容及亚甲基蓝、碘吸附量均随着PVB质量分数的降低而先升高后降低。当PVB质量分数为3.5%时,活化后的高邻位酚醛基活性碳纤维的比表面积为1 409 m2/g,孔容为0.771 cm3/g,亚甲基蓝吸附量为837 mg/g,碘吸附量2 641 mg/g。
FZXB
