(5R)-5-羟基雷公藤硫氰酸基内酯醇的半合成及其体外生物活性研究
2020-12-31郭舜民阙慧卿陈阿虹
李 唯, 郭舜民, 阙慧卿, 陈阿虹, 林 绥
(福建省医学科学研究院 福建省医学测试重点实验室,福建 福州 350001)
雷公藤内酯醇(TP)是雷公藤属植物中的主要活性成分,是一种具有3个环氧基团、具有α,β-不饱和五元内酯环结构,以及14位β朝向羟基的松香烷型二萜化合物。雷公藤内酯醇具有显著和独特的抗炎、免疫抑制、抗肿瘤等药理作用[1-4],其12,13位环氧环可以与通用转录因子TFⅡH上的着色性干皮病B基因亚基(XPB)结合[5],并引起RNA聚合酶Ⅱ的最大亚基Rpb1降解,发挥抗肿瘤作用;TP还可以靶向转化生长因子-β激活激酶1从而阻止巨噬细胞的极化达到抗炎效果[6]。但是由于TP的水溶性差、毒性高等特点限制了其在临床上的应用[7]。经过对TP的结构修饰[8],已经筛选出2个候选药物进入了临床研究阶段,包括(5R)-5-羟基雷公藤内酯醇(LLDT-8)[9]和米内利德(Minnelide)[10]。其中,LLDT-8具有很强的肿瘤和免疫抑制活性,比起先导化合物TP,毒性大大地降低。本课题组在前期研究工作中以雷公藤甲素为原料合成出了雷公藤硫氰酸基内酯醇[11],虽然这个化合物不易转化为TP,但令人惊奇的是该化合物具有很好的抗肿瘤活性。体外细胞毒性试验显示雷公藤硫氰酸基内酯醇具有与TP相当的细胞毒性。在治疗小鼠黑色素瘤的模型中,更以4 mg/(kg·d)的剂量达到50.17%的抑制率,与达卡巴仁(dacarbazine)20 mg/(kg·d)效果相当[12]。相比于TP,雷公藤硫氰酸基内酯醇的12位被硫氰酸基取代,使得12位的亲核能力减弱,不能很好作用于XPB[5],导致毒性降低,有研究表明对XPB抑制作用小的TP衍生物的毒性较小[13-14]。基于前期的研究基础,本研究对已经进入临床研究的(5R)-5-羟基雷公藤内酯醇(LLDT-8)进行结构改造,利用12,13位环氧易受亲核试剂进攻的特点,使用硫氰酸铵作为亲核试剂,进攻12,13位环氧环,得到(5R)-5-羟基雷公藤硫氰酸基内酯醇,并对目标化合物的生物活性包括体外抗肿瘤活性、免疫抑制活性和细胞毒性进行探讨,以期获得高效低毒的LLDT-8衍生物。
1 实 验
1.1 仪器以及试剂
AVANCE NEO 600型核磁共振仪,瑞士Bruker公司;Impact Ⅱ型超高分辨飞行时间质谱仪(HRMS),瑞士Bruker公司。
雷公藤内酯酮,纯度98%,福建省医学科学研究院;硼氢化钠、硫氰酸铵,均为分析纯;二氧化硒,化学纯。二甲基亚砜(DMSO),Merck公司;噻唑蓝(MTT)、杜氏改良Eagle(DMEM)培养基、刀豆蛋白A(ConA),Sigma公司;青链霉素、胰酶消化液,上海碧云天生物技术有限公司;胎牛血清(FBS)、磷酸盐缓冲溶液(PBS),赛默飞公司。人肺腺癌细胞A549和人胚肾细胞293T从中科院上海细胞库购买。小鼠脾脏淋巴细胞分离液购自北京索莱宝生物有限公司。
1.2 目标化合物合成
1.2.1合成路线 雷公藤内酯酮(1)在二氧化硒存在下,二氧六环回流得到(5R)-5-羟基雷公藤内酯酮(2),化合物2使用硼氢化钠还原得到化合物3和4,最终以硫氰酸铵为亲核试剂,分别得到(5R)-5-羟基雷公藤硫氰酸基内酯醇(5)和(5R)-5-羟基表雷公藤硫氰酸基内酯醇(6),合成路线如图1所示,实验收率按以下公式计算:实验收率=(目的产物的实际质量/目的产物的理论质量)×100%。
1.2.2化合物2的合成 称取雷公藤内酯酮(1,300 mg,0.833 mmol)和二氧化硒(365 mg,3.32 mmol),加入到15 mL 1,4-二氧六环溶液中。加热回流24 h后,使用薄层硅胶板(TLC)监测反应原料是否反应完全,展开剂为V(乙酸乙酯) ∶V(石油醚)=1 ∶1.5。待反应液中大部分原料被反应完之后,冷却反应液至室温,用乙酸乙酯萃取3次,有机层用饱和NaCl水溶液洗涤。所得到的有机层再用无水硫酸钠干燥,过滤,浓缩蒸干。浓缩蒸干之后所得到的残余物用四氢呋喃(THF)溶解,再倒入相当于残余物质量1.5倍的硅胶,并使得硅胶充分分散在溶液中,蒸干溶剂得到负载残余物的硅胶粉末,利用柱层析(硅胶粒径50~75 μm,下同)技术对残余物进行纯化,用展开剂(V(二氯甲烷) ∶V(甲醇)=200 ∶1)洗脱,得化合物2。
1.2.3化合物3和化合物4的合成 称取520 mg(5R)-5-羟基雷公藤内酯酮(2)置于10 mL甲醇中,冷却至0 ℃,搅拌下加入208 mg硼氢化钠,反应5 h,固体悬浮在甲醇中,使用TLC监测反应原料是否反应完全,展开剂为V(二氯甲烷) ∶V(甲醇)=100 ∶1。待TLC检测反应发现原料点消失后,向反应液中加入40 mL水,用乙酸乙酯萃取3次,有机层用饱和NaCl水溶液洗涤。回收有机层用无水硫酸钠干燥,过滤,蒸干。浓缩蒸干后的残余物用THF溶解,再倒入相当于残余物质量1.5倍的硅胶,并使得硅胶充分分散在溶液中,蒸干溶剂得到负载残余物的硅胶粉末,利用柱层析技术对残余物进行纯化,用展开剂V(二氯甲烷) ∶V(甲醇)=200 ∶1洗脱,得化合物3(LLDT-8)和化合物4。
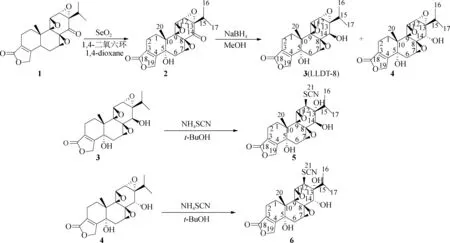
图1 目标化合物的合成路线
1.2.4化合和5和化合物6的合成 化合物3(126 mg,0.33 mmol)加入到15 mL叔丁醇中,80 ℃下搅拌,溶解。随后往反应液中加入硫氰酸铵(376 mg,4.95 mmol),回流反应,使用TLC监测反应原料是否反应完全,展开剂为V(二氯甲烷) ∶V(甲醇)=100 ∶1。待TLC检测反应发现原料点消失后,往反应液中加入40 mL的EtOAc。有机层用饱和NaCl水溶液水洗3次,然后用无水硫酸钠干燥,过滤。减压浓缩,水浴温度55 ℃,得到白色残余物。所得白色残余物用THF溶解,再倒入相当于残余物质量1.5倍的硅胶,并使得硅胶充分分散在溶液中,蒸干溶剂得到负载残余物的硅胶粉末,利用柱层析技术对残余物进行纯化,用展开剂V(二氯甲烷) ∶V(甲醇)=100 ∶1洗脱,收集组分,真空干燥,得化合物5。
化合物4(100 mg,0.26 mmol)加入到15 mL叔丁醇中,80 ℃下搅拌,溶解。随后往反应液中加入硫氰酸铵(296 mg,3.9 mmol),回流反应,TLC监测反应进程,展开剂为V(CH2Cl2) ∶V(MeOH)=100 ∶1。待TCL检测反应发现原料点消失后,往反应液中加入40 mL的EtOAc。有机层用饱和NaCl水溶液水洗3次,再用无水硫酸钠干燥,过滤。减压浓缩,水浴温度55 ℃,得到白色残余物。所得白色残余物用THF溶解,再倒入相当于残余物质量1.5倍的硅胶,并使得硅胶充分分散在溶液中,蒸干溶剂得到负载有残余物的硅胶粉末,利用柱层析技术对残余物进行纯化,用展开剂V(二氯甲烷) ∶V(甲醇)=100 ∶1洗脱,收集组分,真空干燥,得化合物6。
1.3 生物活性测试
1.3.1体外抗肿瘤活性
1.3.1.1细胞培养 人肺腺癌细胞A549用胰酶消化后,10%FBS、1%青链霉素和DMEM完全培养基制成细胞悬浮液,置于37 ℃、5% CO2恒温培养箱培养,1~2天换液一次,3~4天传代一次,待用。
1.3.1.2供试药的配置 将需要测试活性的化合物根据其相对分子质量和质量用少量DMSO溶解,加入PBS将其配成质量浓度为100 g/L的备用溶液。
1.3.1.3细胞处理及MTT法细胞增殖活性检测 取处于对数生长期的人肺腺癌细胞A549细胞,经消化、计数,配制3×107个/L的细胞悬液,在96孔细胞培养板中每孔加入100 μL细胞悬液,各组均设3个平行试验,共接种84孔,置于37 ℃,5% CO2恒温培养箱培养24 h后,加入供试药物(化合物4、5、6和阳性对照LLDT-8(化合物3)),使得药物终质量浓度分别为100、 10、 1、 0.1、 0.01、 0.001 mg/L。加药培养72 h后,每孔加入10 μL 5 g/L的MTT,37 ℃继续孵育4 h,弃去上清液,每孔加入150 μL DMSO,充分混匀,于Bio-Tek酶标仪中测定490 nm处吸光度(OD)值。以100 μL的细胞悬液+20 μL PBS为阴性对照组。100 μL的DMEM完全培养基+20 μL PBS为空白对照组。计算目标化合物的细胞抑制率。
1.3.2免疫抑制实验
1.3.2.1小鼠脾淋巴细胞的制备 小鼠脱颈椎处死,无菌取脾脏放置于筛网上,研磨脾脏使用PBS冲洗筛网,收集细胞悬液,再经过筛网过滤。取离心管,加入与脾脏细胞悬液等量的小鼠脾脏淋巴细胞分离液3 mL。小心吸取细胞悬液加于小鼠脾脏淋巴细胞分离液液面上,离心(800 r/min, 5 min)。用吸管小心吸取环状乳白色淋巴细胞至另一洁净的15 mL离心管中,向离心管中加入10 mL PBS洗涤白膜层细胞,离心(800 r/min, 10 min)。弃去上清液,5 mL的PBS重悬细胞,离心(800 r/min, 5 min)。弃去上清液,收集细胞沉淀,沉淀用10% FBS、1%青链霉素和DMEM完全培养基5 mL悬浮,得到细胞悬液。将得到的脾细胞悬液100 μL加入到96孔培养板中,实验组添加10 μL终质量浓度依次为0.2、 1.0、 2.5、 5、 10、 20 mg/L 的ConA溶液,对照组添加等体积PBS缓冲液。每组设置3个平行试验,然后将培养板置于37 ℃、 5%CO2的培养箱中培养72 h,得出小鼠脾淋巴细胞的分化随着ConA质量浓度的增加呈现先增后减的关系,其最佳适用质量浓度为2.5 mg/L。
1.3.2.2淋巴细胞增殖活性测试 将淋巴细胞置于37 ℃、 5%CO2培养箱进行培养,培养12 h后,以每孔5×103个接种于96 孔板内,设3个平行试验,共78孔,药物(化合物4、5、6和阳性对照LLDT-8(化合物3))终质量浓度分别为100、 10、 1、 0.1、 0.01、 0.001 mg/L。用终质量浓度2.5 mg/L的ConA刺激小鼠脾淋巴细胞的分化,设置空白组和阴性对照组。以100 μL细胞悬液+10 μL ConA+10 μL PBS为阴性对照组;100 μL的DMEM完全培养基+20 μL PBS为空白对照组。加药培养24 h后,每孔加入10 μL 5 g/L MTT,37 ℃继续孵育4 h,弃去上清液,每孔加入150 μL DMSO,充分混匀,于Bio-Tek酶标仪分析490 nm处OD值,计算细胞抑制率和IC50值。
1.3.3细胞毒性实验 取处于对数生长期的人胚肾细胞293T,以每孔3×103个细胞接种于96孔板内,各组均设3个平行试验,置于37 ℃、 5% CO2恒温培养箱培养24 h后,加药处理(化合物4、5、6和阳性对照LLDT-8(化合物3))。药物终质量浓度分别为100、 10、 1、 0.1、 0.01、 0.001 mg/L。加药培养72 h后,每孔加入10 μL 5 g/L MTT,37 ℃继续孵育4 h,弃去上清液,每孔加入150 μL DMSO,充分混匀,于Bio-Tek酶标仪分析490 nm处OD值,计算细胞抑制率和IC50值。
1.4 分析表征
1.4.1目标化合物结构鉴定 DMSO-d6为溶剂,用核磁共振仪进行分析; 用高分辨质谱仪进行 HRMS分析。
1.4.2细胞抑制率和IC50的计算 细胞抑制率和IC50值按以下公式计算:
I=(OD试样-OD阴性对照)/(OD阴性对照-OD空白)×100%
(1)
lgy=Xm-I′[P-(3-Pm-Pn)/4〗
(2)
式中:I—细胞抑制率,%;y—IC50值,mg/L;Xm—设计的最大浓度的对数值;I′—各组最大浓度及其相邻浓度之商的对数值;P—各组生长抑制率之和,%;Pm—最大阳性反应率,%;Pn—最小阳性反应率,%。
2 结果与讨论
2.1 目标化合物的结构表征
制备的目标化合物共有5种,各化合物的C原子编号如图1所示,通过NMR及高分辨质谱(HRMS),对化合物进行了表征。
化合物2:C20H22O7,鳞状白色固体,258 mg,收率83%。1H NMR(400 MHz, DMSO-d6)δ: 4.88(m, 2H, 19-H), 4.11(d,J=2.9 Hz, 1H, 11-H), 4.08(d,J=2.9 Hz, 1H, 12-H), 3.42(d,J=4.4 Hz, 1H, 7-H), 2.22~2.28(m, 1H, 15-H), 1.96~2.20(峰形重叠, 4H, 2-H, 6-H), 1.87~1.79(m, 1H, 1-H), 1.12~1.05(m, 1H, 1-H), 0.91(s, 3H, 20-H), 0.88(d,J=6.9 Hz, 3H, 16-H), 0.78(d,J=6.9 Hz, 3H, 17-H);HRMS(电子轰击离子源): [M+H]+375.144 4。化合物2的表征结果与文献[15]报道的数据一致,说明结构和文献所报道的结构一致,确定为(5R)-5-羟基雷公藤内酯酮。化合物2使Kedd’s试剂显紫红色,这是因为在碱性醇溶液中,内酯的活性亚甲基与硝基苯母核缩合氧化生成醌式结构,产生紫红色反应,因此雷公藤内酯醇衍生物具有特殊的α,β-不饱和五元内酯环结构的特性。
化合物3:C20H24O7,鳞状白色固体,126 mg,收率24%。1H NMR (600 MHz, DMSO-d6)δ: 5.31(s, 1H, 5-OH), 5.30(d,J=7.4 Hz, 1H, 14-OH), 4.94~4.84(m, 2H, 19-H), 4.15(d,J=7.4 Hz, 1H, 14-H), 3.73(d,J=3.2 Hz, 1H, 11-H), 3.59(d,J=5.3 Hz, 1H, 7-H), 3.45(d,J=3.1 Hz, 1H, 12-H), 2.31~2.20(m, 1H, 15-H), 2.20~2.07(峰形重叠,3H, 2-H, 6-H), 2.05~1.94(m, 1H, 6-H), 1.84~1.74(m, 1H, 1-H), 1.08~1.02(m, 1H, 1-H), 1.00(s, 3H, 20-H), 0.98(d,J=6.8 Hz, 3H, 17-H), 0.71(d,J=7.0 Hz, 3H, 16-H);13C NMR(151 MHz, DMSO-d6)δ: 173.78(C-18), 163.36(C- 4), 124.58(C-3), 70.24(C-14), 69.18(C-5), 66.12(C-19), 65.92(C-13), 64.37(C-9), 62.60(C-8), 57.11(C-7), 54.03(C-11), 52.78(C-12), 30.68(C- 6), 27.37(C-15), 24.19(C-1), 19.58(C-17), 17.38(C-16), 16.77(C-2), 16.20(C-20)。HRMS(电子轰击离子源):[M+H]+377.160 0。与文献[16]报道数据比对,确定化合物3为(5R)-5-羟基雷公藤内酯醇(LLDT-8)。
化合物4:C20H24O7,鳞状白色固体,330 mg,收率63%。1H NMR(600 MHz, DMSO-d6)δ: 5.34(s, 1H, 5-OH), 4.90~4.85(m, 2H, 19-H), 4.60(d,J=7.3 Hz, 1H, 14-OH), 3.74(d,J=3.1 Hz, 1H, 11-H), 3.54(d,J=2.5 Hz, 1H, 12-H), 3.38(d,J=7.4 Hz, 1H, 14-H), 3.35(d,J=5.3 Hz, 1H, 7-H), 2.21~2.05(峰形重叠, 4H, 2-H, 6-H), 2.04~1.94(m, 1H, 2-H), 1.83~1.71 (m, 1H, 1-H), 1.05(dd,J=12.5, 5.6 Hz, 1H, 1-H), 1.00(s, 3H, 20-H), 0.90(d,J=6.9 Hz, 3H, 17-H), 0.77(d,J=6.9 Hz, 3H, 16-H);13C NMR(151 MHz, DMSO-d6)δ: 173.76(C-18), 163.23(C- 4), 124.61(C-3), 71.54(C-14), 70.18(C-5), 69.13(C-19), 64.43(C-13), 63.12(C-9), 61.87(C-8), 59.25(C-7), 56.41(C-11), 54.52(C-12), 40.26(C-10), 30.80(C- 6), 27.85(C-15), 23.62(C-1), 18.08(C-17), 17.29(C-16), 17.11(C-2), 16.81(C-20)。HRMS(电子轰击离子源): [M+H]+377.345 0。与文献[16]报道的数据比对,确定化合物4为(5R)-5-羟基表雷公藤内酯醇。Kedd’s试剂显紫红色。
化合物5:C21H25NO7S,(5R)-5-羟基雷公藤硫氰酸基内酯醇,白色粉末,82 mg,收率57%。m.p.:248~250 ℃。1H NMR(600 MHz, DMSO-d6)δ: 6.20(s, 1H, 5-OH), 5.25(d,J=6.6 Hz, 1H, 14-OH), 5.01~4.82 (m, 2H, 19-H), 4.92 (s, 1H, 13-OH), 4.06 (d,J=5.5 Hz, 1H, 12-H), 3.88 (d,J=6.6 Hz, 1H, 7-H), 3.84 (d,J=5.5 Hz, 1H, 14-H), 3.69 (d,J=6.0 Hz, 1H, 11-H), 2.26 (d,J=15.6 Hz, 1H, 2-H), 2.20~2.12 (m, 1H, 2-H), 2.20~2.12 (m, 1H, 6-H), 2.11~2.05 (m, 1H, 15-H), 2.05~1.97 (m, 1H, 6-H), 1.83~1.65 (m, 1H, 1-H), 1.17~1.11 (m, 1H, 1-H), 1.09 (d,J=5.2 Hz, 3H, 16-H), 1.08 (d,J=4.9 Hz, 3H, 17-H), 1.00 (s, 3H, 20-H);13C NMR(151 MHz, DMSO-d6)δ: 173.49(C-18), 160.95(C-3), 126.09(C- 4), 112.65(C-21), 78.99(C-13), 71.48(C-5), 69.43(C-19), 68.66(C-9), 65.88(C-7), 62.08(C-8), 59.33(C-14), 53.34(C-11), 49.54(C-12), 40.00(C-10), 33.52(C-15), 29.96(C-2), 25.27(C-1), 18.40(C-17), 17.26(C- 6), 16.77(C-16), 16.75(C-20);HRMS(电子轰击离子源): [M+Na]+458.124 4。Kedd’s试剂显紫红色。
化合物6:C21H25NO7S,(5R)-5-羟基表雷公藤硫氰酸基内酯醇,白色粉末,71 mg,收率61%。m.p.:258~260 ℃。1H NMR(600 MHz, DMSO-d6)δ: 6.37(s, 1H, 5-OH), 5.65(d,J=4.4 Hz, 1H, 14-OH), 5.07~4.85 (m, 2H, 19-H), 4.79 (d,J=1.7 Hz, 1H, 13-OH), 3.92 (d,J=5.9 Hz, 1H, 11-H), 3.81 (d,J=5.8 Hz, 1H, 12-H), 3.37 (d,J=5.9 Hz, 1H, 7-H), 3.11 (d,J=4.4 Hz, 1H, 14-H), 2.33~2.22 (m, 2H, 2-H), 2.20~2.12 (m, 1H, 15-H), 2.20~2.12 (m, 1H, 6-H),2.07~1.93 (m, 1H, 6-H), 1.76~1.71(m, 1H, 1-H), 1.37~1.14 (m, 1H, 1-H), 1.02 (d,J=6.8 Hz, 3H, 16-H), 0.98 (s, 3H, 20-H), 0.85 (d,J=6.7 Hz, 3H, 17-H);13C NMR (151 MHz, DMSO-d6)δ: 173.48(C-18), 160.86(C-3), 126.22(C- 4), 115.71(C-21), 77.46(C-13), 74.84(C-14), 71.36(C-5), 69.39(C-19), 66.02(C-9), 60.09(C-8), 59.24(C-7), 57.59(C-11), 51.13(C-12), 40.00(C-10),29.98(C-2), 29.30(C-15), 24.83(C-1), 17.22(C-6), 16.93(C-20), 16.52(C-17), 15.96(C-16);HRMS(电子轰击离子源): [M+Na]+458.123 9。Kedd’s试剂显紫红色。
化合物3和4的1H NMR数据显示在低场区存在2个羟基氢的信号(5-OH和14-OH)。14位羟基会受到相邻氢的影响产生裂分,因此判断化合物3和4中δ5.30和δ4.60的信号分别对应14位羟基氢的信号。对比文献[16]报道数据发现,当14位羟基为β朝向时,化学位移会更加偏向于低场,因此判断化合物3为LLDT-8,即14位羟基朝向为β朝向。
对比化合物3与化合物5的谱图,以及对比化合物4与化合物6的图谱后发现一个新的羟基信号。在异核多键相关谱(HMBC)中该羟基氢信号与13、 14和15位氢有远程耦合,说明在反应中LLDT-8的12、 13位环氧环发生开环,形成了一个新的羟基。高分辨质谱(HRMS)分析确定的分子式说明化合物5在12位引入了硫氰酸基。基于奥氏核效应交换相关谱(NOESY),化合物5的14位羟基氢与16、 17位甲基上的氢有空间上的相关,说明14位羟基氢为β朝向,确认结构为(5R)-5-羟基雷公藤硫氰酸基内酯醇。而化合物6的NOESY中没有出现与16、 17位甲基上的氢的相关信号,说明14位羟基氢为α朝向,确定化合物6为化合物5的差向异构体。
2.2 目标化合物的生物活性评价
2.2.1体外抗肿瘤活性 合成得到目标化合物之后,对目标化合物进行了抗肿瘤细胞增殖活性检测,待测化合物相对于对照化合物3(LLDT-8)对肿瘤细胞抑制率和IC50值如表1所示。LLDT-8的差向异构体化合物4的抗肿瘤活性与LLDT-8相比下降明显,说明14-OH为α朝向不利于与生物体内的相关靶点作用,使得抗肿瘤活性下降。化合物5和化合物6在质量浓度小于或者等于1 mg/L下时对A549细胞抑制作用较弱。当质量浓度10 mg/L时,化合物5对A549的抑制率达到69.95%,与LLDT-8在质量浓度10 mg/L下对A549的抑制率相当,说明10 mg/L的质量浓度下化合物5具有很强抑制肿瘤细胞A549生长的作用。而在质量浓度10 mg/L下化合物6对A549细胞的抑制率只有16.60%,表现出较弱的抑制作用。化合物5和化合物6是一对差向异构体,14位羟基为β朝向的化合物5的抗肿瘤活性(IC506.78 mg/L)要优于α朝向的抗肿瘤活性(IC5035.54 mg/L)。
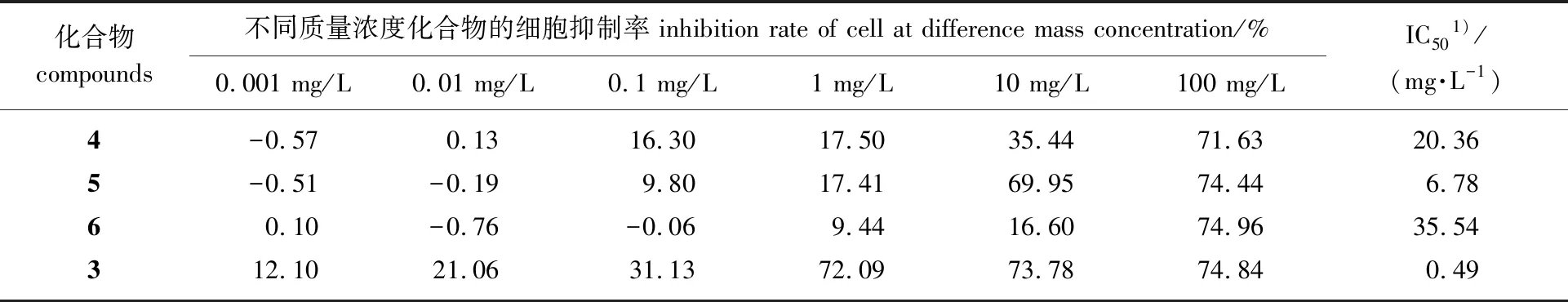
表1 目标化合物的抗肿瘤(A549)活性
2.2.2免疫抑制活性 由于雷公藤内酯醇衍生物具有广泛的生物学活性,且LLDT-8正在进行活动性类风湿性关节炎的治疗,所以对目标化合物进行了免疫抑制活性测试,探讨了待测化合物相对于对照LLDT-8(化合物3)对ConA诱导的脾淋巴细胞体外增殖的抑制作用,结果如表2所示。
实验结果表明:14-OH为α朝向的化合物4和化合物6比起LLDT-8免疫抑制活性都有大幅下降,化合物4和6的IC50值分别为13.78和26.33 mg/L,LLDT-8的IC50值为1.58 mg/L;化合物5在质量浓度10 mg/L 下对淋巴细胞的增殖抑制率为53.37%,低于阳性对照LLDT-8在相同质量浓度下的抑制率71.23%,另外化合物5对淋巴细胞的IC50值为17.36 mg/L,比起阳性对照(1.58 mg/L)也有显著的下降。
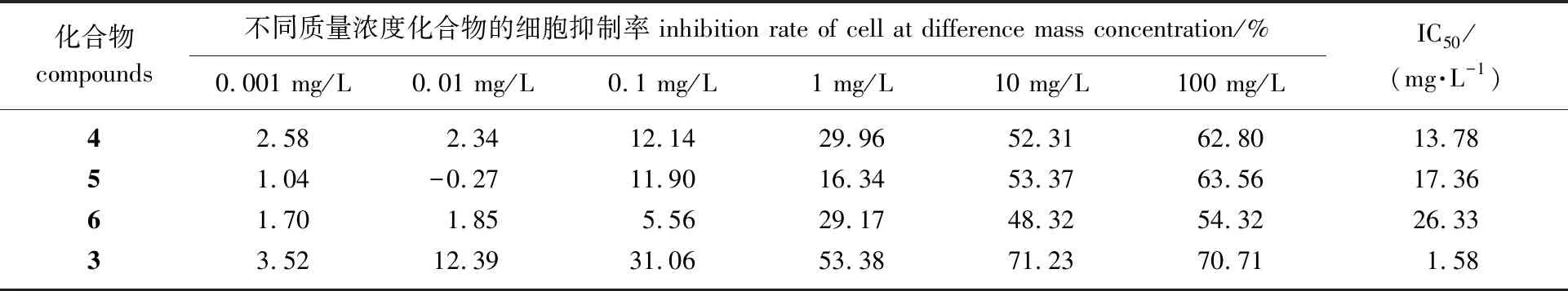
表2 目标化合物的免疫抑制活性
2.2.3细胞毒性实验 待测化合物相对于对照LLDT-8(化合物3)对正常细胞(人胚肾293T细胞)抑制率,即细胞毒性数据如表3所示。从细胞毒性实验来看,化合物5在质量浓度10 mg/L下对A549细胞的抑制率就能达到69.95%,而在质量相同浓度下对293T细胞的抑制率只有54.41%,说明化合物5对肿瘤细胞的毒性比对正常细胞强。当在质量浓度100 mg/L下,化合物5对293T细胞的抑制率60.04%,而LLDT-8的抑制率为70.63%,说明化合物5对正常细胞的毒性比对照LLDT-8有一定的降低。化合物4和6的IC50值分别10.65 mg/L和18.12 mg/L,都高于阳性对照对293T细胞的IC50值(1.93 mg/L),说明化合物4和6的生物活性下降的同时,细胞毒性也降低了。
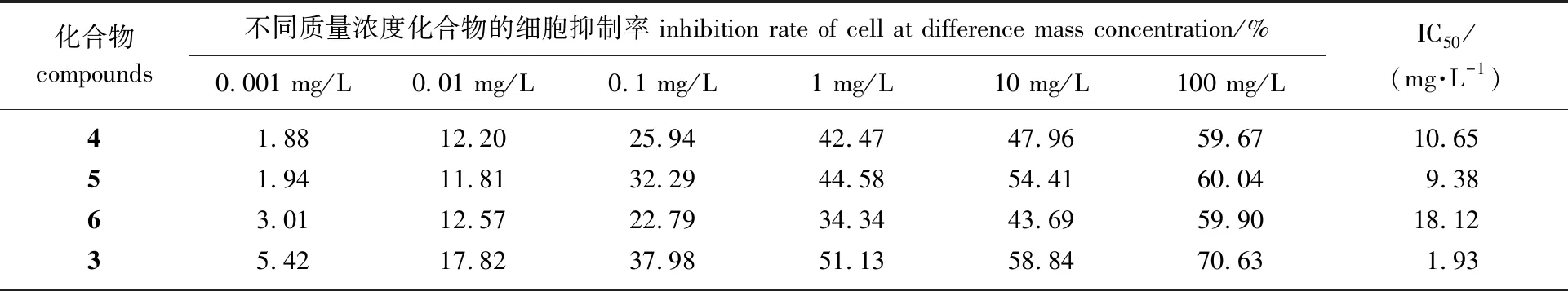
表3 目标化合物的对293T细胞的抑制活性
3 结 论
以雷公藤内酯酮为原料首先合成了(5R)-5-羟基雷公藤内酯酮(2),然后合成了LLDT-8(3)及其差向异构体(4),然后使用硫氰酸铵进攻LLDT-8及其差向异构体的12和13位得到(5R)-5-羟基雷公藤硫氰酸基内酯醇(5)及其差向异构体(6)。利用NMR和高分辨质谱对化合物进行了表征,确证合成了目标化合物。对目标化合物的生物活性进行初步研究表明:14-OH为α朝向的LLDT-8差向异构体(4)的生物活性相比LLDT-8减弱;12、 13位环氧基团开环之后的化合物5在质量浓度为10 mg/L对人肺腺癌细胞A549的抑制率已经与相同质量浓度下的阳性对照LLDT-8对A549的抑制率相当,说明化合物5具有很强的抗肿瘤活性;化合物5对淋巴细胞增殖抑制活性与阳性对照LLDT-8相比有一定程度降低;安全性提高的同时一般也伴随着活性的降低,需要进一步发现更多提高治疗窗口的雷公藤内酯醇衍生物。
