RP-HPLC混标加样增量法快速测定食品中的阿斯巴甜和阿力甜
2020-12-29高向阳
高向阳,张 芳
(郑州科技学院食品科学与工程学院,郑州市食品安全快速检测重点实验室,河南 郑州 450064)
阿斯巴甜即天门冬酰苯丙氨酸甲酯,是一种天然功能性低聚糖,甜度是蔗糖的200 倍。在pH 3~5的溶液中稳定,高pH值或高温下会水解而导致甜味丧失[1-3]。阿力甜又称为天胺甜精,甜度为蔗糖的2 000 倍以上,极易溶于水或溶于含羟基的溶剂中。室温下,阿力甜在pH 4的条件下能稳定2 a,在pH 5~8的水溶液中能稳定 4 a以上[4-5]。阿斯巴甜和阿力甜复合使用可以协同增效、降低成本、改善口感、提高甜味的稳定性,不但甜度高、味质好,且储存期长、无热量,是常用的食品甜味剂。但若使用过量可能会出现人体肝脏被破坏、神经系统紊乱、致癌等不良影响[6-10]。
目前,食品中测定阿斯巴甜和阿力甜的方法有高效液相色谱法[11-18]、超高效液相色谱法[19]、反相高效液相色谱法[20]、液相色谱-串联质谱法[21-23]、超高效液相色谱-串联质谱法[24-26]、离子色谱法[27]等,而且常用外标法、内标法或归一化法进行定量分析。
外标法需要绘制标准曲线,标准液和待测液要分开进行测定,不能实现同时、同步分析,容易受到仪器条件以及环境因素的影响。内标法需要先用标准液求得各待测组分和内标物的相对校正因子,代入相应公式计算结果。对复杂样品,有时很难找到合适的内标物,也无法确定样品中是否不含所用的内标物,很难及时开展工作。归一化法要求样品中所有组分全部出峰、并产生测定信号,样品中各组分的相对含量之和应为100%。要求知道各组分的校正因子,操作较为繁琐。这些测定方法操作繁杂,需测定空白溶液、绘制标准曲线或进行复杂计算,工作效率较低。
混标加样增量法是一种新型分析技术,克服了上述分析方法的不足,只需取2 份完全相同的阿斯巴甜和阿力甜混合标准液,依次加入不同体积的试液,混匀后在同一条件下进行测定。以同一保留时间下组分色谱峰信号是否有增加进行定性分析,以混合标准液加样后色谱峰信号值代入公式进行定量分析,无需绘制标准曲线和测定空白溶液,具有成本低、工作效率高、测定速度快、干扰少、准确度高、适用范围广等突出优点。
1 材料与方法
1.1 材料与试剂
可口可乐、菊花晶、酸奶、浓缩果汁、巧克力制品 市售。
甲醇(色谱纯)、乙醇(优级纯)、阿力甜标准品(C14H25N3O4S,CAS号:80863-62-3,纯度为99.2%) 上海安谱实验科技股份有限公司;阿斯巴甜标准品(C14H18N2O5,CAS号:22839-47-0,纯度为95%) BePune实验科技有限公司。
1.2 仪器与设备
A100250多管涡旋混合仪 月旭科技股份有限公司; MS150/MS205DU电子天平(十万分之一) 长沙市秋龙仪器设备有限公司;Dynamica V18R台式冷冻离心机 北京五洲东方科技发展有限公司;SB25-12DTD型超声波清洗机 宁波新芝生物科技股份有限 公司;Utimate3000(2017LH131)高效液相色谱仪、Hypersil GOLD C18色谱柱(150 mm×4.6 mm) 美国赛默飞科技股份有限公司。
1.3 方法
1.3.1 标准混合液的制备
乙醇溶液:取40 mL乙醇,与20 mL水均匀混合;80%甲醇溶液:取80 mL甲醇,用水定容到100 mL容量瓶中;50%甲醇溶液:取50 mL甲醇,用水定容到100 mL容量瓶中。
阿斯巴甜和阿力甜标准储备液的配制(0.50 mg/mL): 称取阿斯巴甜0.026 32 g(精确至0.000 1 g),称取阿力甜0.025 20 g(精确至0.000 1 g),用水将阿斯巴甜和阿力甜分别溶解至50 mL容量瓶内并定容至刻度,置于4 ℃左右的冰箱中保存,有效期为90 d。
1.3.2 试液的制备和测定
阿斯巴甜和阿力甜混合标准工作液系列的配制:将阿斯巴甜和阿力甜标准储备液用水逐级稀释,并将2 种标准溶液等浓度等体积混合。分别制得50.00、45.00、40.00、35.00、30.00、25.00、20.00、10.00、5.00、2.50、1.00、0.50 µg/mL阿斯巴甜和阿力甜的混合标准溶液,置于4 ℃左右冰箱保存,有效期为30 d。
1.3.2.1 碳酸饮料、固体饮料样品处理
称取约5.000 0 g(精确到0.001 g)碳酸饮料试样于50 mL烧杯中,超声振荡除去二氧化碳,转入25 mL容量瓶中,用水定容,备用。
称取约1.000 0 g(精确到0.001 g)固体饮料试样于50 mL烧杯中,加10 mL水,超声振荡提取20 min,移入25 mL容量瓶中;再向烧杯中加入10 mL水超声振荡提取10 min,将2 次提取液移入同一个25 mL的容量瓶中,用水定容,备用。
将上述容量瓶的液体分别4 000 r/min离心5 min,取上清液用0.45 µm的水系滤膜过滤后得待测试液,用于液相色谱分析。
1.3.2.2 酸奶、乳饮料样品处理
称取5.000 0 g(精确到0.001 g)酸奶试样于50 mL离心管中,加入10 mL乙醇,沉淀酸奶试样中的蛋白质,盖紧盖子;上下轻轻颠倒离心管5 次(不能振摇),将离心管混匀涡旋10 s后,静置1 min,4 000 r/min离心5 min,取上清液滤入25 mL容量瓶中,滤渣用8 mL乙醇溶液洗涤后再离心,离心后的上清液移入同一个容量瓶,用乙醇溶液定容至25 mL,混匀,用0.45 μm有机系滤膜过滤后得待测试液,用于液相色谱分析。
1.3.3 色谱分析
取2 份完全相同的混合标准液于离心管A和B中,离心管A中加入体积为VA的待测试液,离心管B中加入体积为VB的待测试液,其中VB>VA;在同一色谱条件下,对离心管A中混合液进行色谱分析,得出阿斯巴甜保留时间tA1及对应的出峰信号hA1,阿力甜保留时间tA2及对应的出峰信号hA2;对离心管B中混合液进行色谱分析得出阿斯巴甜的保留时间tB1及对应的出峰信号hB1,阿力甜的保留时间tB2及对应的出峰信号hB2。当tA1=tB1,hB1-hA1>0,则待测食品中含有阿斯巴甜;当tA2=tB2,hB2-hA2>0,则待测食品中含有阿力甜。
1.3.4 测定原理和公式
在一定条件下测定时,测定信号与待测组分的量呈正比,因此有:

在完全相同的条件下测定时,比例常数k相同, 式(1)除以式(2)有:

式中:ρ 为试液中阿斯巴甜或阿力甜的质量浓 度/(μg/μL);ms为混合标准液中阿斯巴甜或阿力甜的质量/μg;VA为离心管A中加入试液的体积/μL;VB为离心管B中加入试液的体积/μL;mA和mB分别为试液加入A管和B管中的组分质量/μg;V样为总的试液体积/μL; m样为总样品质量/g;m为总样品中阿斯巴甜或阿力甜的质量/μg;hA、hB分别为A、B离心管中阿斯巴甜或阿力甜测得峰高信号值。
2 结果与分析
2.1 条件的选择
2.1.1 色谱流动相的选择
用不同比例梯度的甲醇和水作为流动相,阿斯巴甜和阿力甜能很好得到分离,最佳流动相为甲醇-水(40∶60,V/V),流速为0.8 mL/min。
2.1.2 波长的选择
基于阿斯巴甜和阿力甜在紫外光区的检测灵敏度,选择200 nm波长检测,确定阿斯巴甜和阿力甜的保留时间如图1所示,结果表明,阿斯巴甜与阿力甜出峰较好。

图1 阿斯巴甜和阿力甜混合标准溶液的保留时间色谱图Fig. 1 Chromatograms showing retention times of mixed standard solution of aspartame and alitame
2.2 加样增量法色谱图比较分析
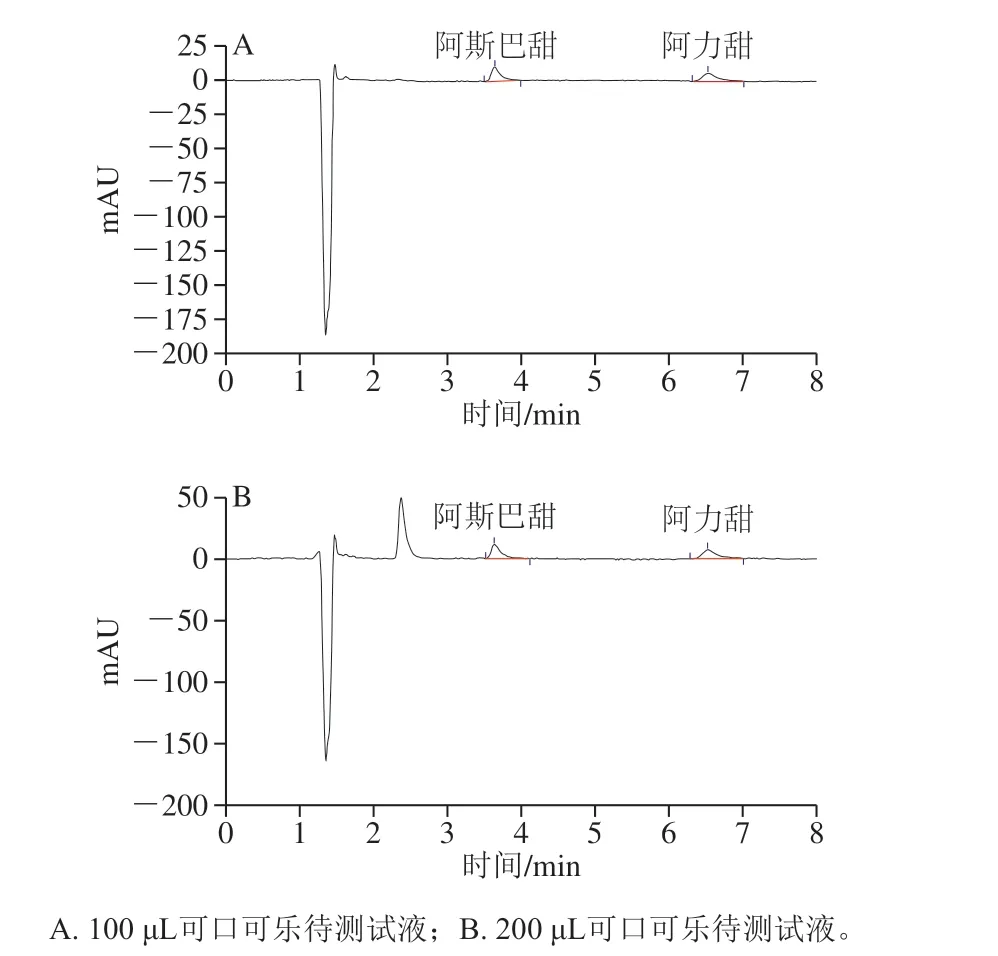
图2 阿斯巴甜和阿力甜混合标准液加不同量可口可乐色谱图Fig. 2 Chromatograms of in aspartame and alitame in Coca-Cola added with different amounts of mixed standard solution
取2 个相同的离心管A、B,各加10 μg/mL阿斯巴甜和阿力甜混合标准液50 μL,再向离心管A中加入100 μL可口可乐待测试液,向离心管B中加入200 μL可口可乐待测试液,均定容至1.00 mL,混匀后进行色谱分析。由图2 可知,阿斯巴甜和阿力甜在各自保留时间下,加入不同体积的可口可乐试液后,各自的峰高均有增加,保留时间允许的误差范围内tA1=tB1,hB1-hA1>0,则可口可乐中含有阿斯巴甜,tA2=tB2,hB2-hA2>0,则可口可乐中含有阿力甜,以此进行定性分析。
由图2A可得,阿斯巴甜保留时间tA1为3.643 min,峰高hA1为10.188 mAU,阿力甜保留时间tA2为6.527 min,峰高hA2为6.260 mAU;如图2B所示:阿斯巴甜保留时间tB1为3.633 min,峰高hB1为11.464 mAU,阿力甜保留时间tB2为6.520 min,峰高hB2为7.163 mAU。根据色谱图的峰高,依据式(3)、(4)可对样品中的组分进行定量分析。

表1 样品的测定结果Table 1 Results obtained from determination of aspartame and alitame in real samples
2.3 加样增量法测定结果及精密度

由表1可知,5 种试样测定的相对标准偏差(relative standard deviation,RSD)均小于5%。由色谱峰高值,可简便、快速计算出食品中待测组分的含量,也可用峰面积数据代入相应公式计算。
2.4 样品的检出限和定量限结果

表2 样品的检出限和定量限Table 2 Detection limits and quantitation limits for real samples
根据表1,按3 倍标准差计算检出限,按10 倍标准差计算定量限,见表2。由GB 5009.263—2016《食品中阿斯巴甜和阿力甜的测定》的检出限和定量限可知[28],碳酸饮料、液态乳饮料中的阿斯巴甜和阿力甜的检出限为 1 mg/kg,定量限为3 mg/kg,巧克力制品、水果及其制品、固体饮料及其制品中的阿斯巴甜和阿力甜的检出限为5 mg/kg,定量限为15 mg/kg。该方法的检出限和定量限均低于国家食品安全标准中的检出限和定量限。
2.5 加标回收率结果
以酸奶测定加标回收率,分别对样品进行3 个水平的加标回收,每个水平做6 次平行测定,检验无可疑值后,取峰高平均值计算,见表3。

表3 样品的加标回收率Table 3 Recoveries for spiked samples
由表3 可知, 酸奶中阿斯巴甜的回收率为90.5%~98.3%,阿力甜的回收率为93.2%~95.8%。由于各组分在色谱柱中得到了充分分离,互不干扰,尤其适用于多组分的色谱定性和定量分析,由各组分依次出现的色谱峰信号值,可快速计算各组分含量和回收率。
2.6 国标法对比实验
2.6.1 国标法标准曲线的回归方程
按国标法规定的波长和色谱条件,以5 0.0 0、45.00、40.00、35.00、30.00、25.00、20.00、10.00、5.00、2.50、1.00 μg/mL和0.50 μg/mL的标准混合使用液各进样5 μL测定,以组分质量浓度为横坐标,以峰高为纵坐标绘制标准曲线,得出阿斯巴甜的回归直线方程为Y=4.692 1x-1.464 9,阿力甜的回归直线方程为Y=2.883 6x-1.194 4,相关系数r均为0.999 9。
2.6.2 国标法测定结果

表4 国标法测定结果Table 4 Comparison of results obtained from determination of aspartame and alitame in real samples by the proposed method and the national standard method
按国标法操作步骤,各样品均进行6 次平行测定,检验无可疑值后,取平均值报告,见表4。国标法采用外标法绘制标准曲线并测定样品中待测物质的含量。结果表明,阿斯巴甜和阿力甜含量最低的为酸奶,最高的为巧克力,RSD前者为0.7%~4.8%,后者为0.48 %~2.2%。
2.7 显著性检验结果

表5 本实验方法与国标法对照测定结果Table 5 Comparative statistical analysis of aspartame and alitame contents of yogurt determined by the proposed method and the national standard method
以酸奶为样品,样品前处理依据GB 5009.263—2016,用该法和国标法进行对照实验,各样品进行6 次平行测定,结果如表5所示。由表5数据和参考文献[29]可知:F值<F0.05=5.05,表明混标加样增量法与国标法之间不存在显著的偶然误差;t值<t0.05=2.23,表明混标加样增量法与国标法间也不存在显著的系统误差,结论的置信度为95%。
3 结 论
方法所用标准溶液、待测液和试剂量少,成本低,操作简单,无需绘制标准曲线、无需测定空白和复杂计算,可快速获得分析结果。实现了标准溶液和试液在同溶液背景、同体系、同方法、同仪器、同条件、同步操作下进行同时定性、定量分析,准确度得到保障和提升,有一定的创新性。由于各组分在色谱柱中得到了充分的分离,相互之间不干扰,尤其适用于色谱多组分的同时定性和定量分析。与国标法对照,用于测定食品中的阿斯巴甜和阿力甜,经检验无显著性差异。
该法不仅适用于液相色谱法,也适用于气相色谱、离子色谱、超临界流体色谱、毛细管电泳色谱法,以及化学发光、分子荧光、磷光分析法、生物发光分析法、原子发射光谱、原子吸收光谱、原子荧光光谱和紫外、可见分子吸收光谱等诸多分析技术。不仅适用于阿斯巴甜和阿力甜的测定,也适用于其他物质诸多组分的测定。根据所用方法不同,测定信号可以是色谱峰峰高、色谱峰峰面积、相对发光强度、吸光度等,可行性和推广应用领域十分广泛。
