联苯三唑醇磁性分子印迹聚合物的制备及其在食品检测中的应用
2020-12-29田景升张展展秦思楠高文惠
田景升,张展展,秦思楠,高文惠
(河北科技大学生物科学与工程学院,河北 石家庄 050000)
联苯三唑醇是一种常见的广谱、高效、低毒、内吸性三唑类杀菌剂[1],其结构式如图1所示。三唑类农药不仅可以通过破坏细胞膜结构,致使膜渗漏加剧,从而铲除病原菌;而且可以缓解植物叶绿素分解、提高内源脱落酸分解以达到治疗植物的目的;更可通过种子内吸进入植株根系,长时间存留在种子区或根围土壤中,进而对植物起保护作用,它主要用于防治果树黑星病、腐烂病、麦叶穿孔病等[2-6]。但是由于施药不合理及超量使用,使三唑类农药在食品中残留甚至超标,进而对人体健康产生危害,造成较为严重的食品安全问题[7],因此建立相关的检测方法具有重要意义。目前针对三唑类农药的检测方法主要有高效液相色谱(high performance liquid chromatography,HPLC)法[8-10]、气相色谱法[11]、色谱-质谱联用技术[12-17]等。近年来,采用分子印迹技术制备联苯三唑醇印迹聚合物对样品进行前处理已有报道[18-20],但对其磁性分子印迹聚合物(magnetic molecularly imprinted polymer,MMIP)的研究鲜见报道。
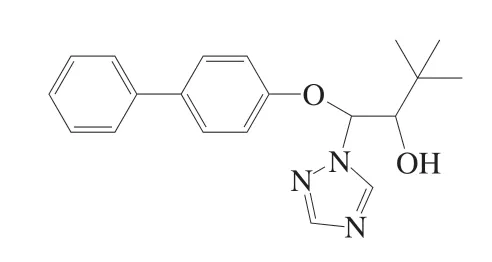
图1 联苯三唑醇的结构式Fig. 1 Structure of bitertanol
磁性聚合物微球是近些年来分子印迹技术重点研究的内容之一,磁性聚合物微球可以在外磁场的作用下迅速与溶液分离,无需离心或者过滤即可达到主动识别和分离[21-23];与此同时,由于增大了聚合物微球的比表面积,使得聚合物的网络中存在很多印迹空穴,所以该材料具有高度的识别选择性[24-26];目前磁性聚合物微球已在物质的提取与分离[27]、特异性识别[28-29]、食品安全检测[30]等 方面得到应用。本实验以Fe3O4作为磁核,通过包覆SiO2并在其表面接枝硅烷偶联剂KH570(甲基丙烯酰氧基丙基三甲氧基硅烷)得到磁性载体,然后采用表面印迹法,以联苯三唑醇为模板分子,制备具有核-壳结构的MMIP。考察乙腈用量、聚合温度及充氮气时间等条件对印迹聚合物印迹效果的影响。以MMIP为固相萃取材料富集和净化样品,并结合HPLC对食品样品中三唑类农药进行检测。
1 材料与方法
1.1 材料与试剂
小米、小麦、白菜、梨、苹果均为市售。
联苯三唑醇(纯度99%) 江苏常州市武进振华化工厂;FeCl2·4H2O(纯度99.7%)、FeCl3·6H2O(纯 度99%) 上海市宝山区合众化工厂;硅酸四乙酯(纯度99%)、硅烷偶联剂(KH-570,纯度97%) 篮西科技有限公司;α-甲基丙烯酸酯(methacrylic acid,MAA,分析纯)、2,2’-二偶氮异丁腈(azobisisobutyronitrile,AIBN,分析纯) 常州畅通化工有限公司;二甲基丙烯酸乙二醇酯(ethylene dimethacrylate,EDMA, 分析纯) 四川海诺威科技有限公司;氨水、乙酸、乙腈、甲醇、无水乙醇、异丙醇(均为分析纯) 成都欣正通化工有限公司;实验用水为超纯水。
1.2 仪器与设备
LC-1260型HPLC仪 美国安捷伦科技有限公司; SHZ-82A型恒温水浴振荡器 上海跃进医疗器械厂;HH-4型恒温水浴锅 上海晖创化学仪器有限公司;DZF-6320型真空干燥箱 四川中浪科技有限公司;KH5200型超声波清洗器 上海沪粤明科学仪器有限公司;SHB-3A 型循环水式真空泵 陕西太康生物科技有限公司;TENSOR37型傅里叶变换红外光谱仪 德国Bruker 公司;JEM-2100型透射电子显微镜 日本Hitachi公司。
1.3 方法
1.3.1 磁核载体的制备及改性
1.3.1.1 共沉淀法制备Fe3O4
分别称取4.72 g FeCl3·6H2O和1.72 g FeCl2·4H2O,放入250 mL的三口烧瓶中,然后向其加入80 mL水,在通入氮气和水浴温度为80 ℃的反应条件下,逐滴加入10 mL 30%氨水溶液,800 r/min搅拌条件下,熟化30 min,再使用磁铁将所得产物分离,然后使用除氧水将产物洗至中性,无水乙醇洗涤3 次后,于50 ℃真空干燥24 h,即得实验用Fe3O4。
1.3.1.2 SiO2包覆Fe3O4
称取300 mg Fe3O4,置于250 mL三口瓶中,分别加入4 mL水和50 mL异丙醇,超声混匀20 min,800 r/min连续搅拌条件下,加入5 mL 30%氨水,再将2 mL硅酸四乙酯逐滴加入,在室温条件下反应12 h,然后用磁铁将产物分离,用水将产物洗涤至中性,再用无水乙醇洗涤3 次,随后在50 ℃真空干燥24 h,得到实验用Fe3O4@SiO2。
1.3.1.3 Fe3O4@SiO2接枝KH570
称取200 mg Fe3O4@SiO2,置于250 mL三口瓶中,加入50 mL甲醇,超声混匀。550 r/min连续搅拌条件下,逐滴加入3 mL KH570,在室温条件下反应24 h,用磁铁将产物分离后,用无水乙醇和水交替洗涤产物数次,并在50 ℃真空干燥24 h,得到实验用Fe3O4@SiO2-KH570。
1.3.2 MMIP的制备
取0.2 mmol联苯三唑醇和0.8 mmol MAA溶于乙腈中,超声振荡30 min,放入冰箱内预聚合6 h,使MAA与联苯三唑醇充分作用,随后加入100 mg Fe3O4@SiO2-KH570、1.5 mL EDMA和40 mg AIBN,对其进行超声脱气,充氮气10 min使其内部无氧,密封后150 r/min恒温振荡24 h;并用甲醇-乙酸(9∶1,V/V)对磁分离得到的聚合物产物进行洗涤,以去除模板分子和未反应的功能单体、交联剂等,再用甲醇洗涤去除乙酸。60 ℃真空干燥(0.04 MPa)至质量恒定后得到MMIP,放入干燥器内。在不加联苯三唑醇的条件下,按上述制备过程制得磁性非分子印迹聚合物(magneticnon-imprinted polymer,MNIP)。
1.3.3 不同聚合物吸附性能
在具塞玻璃管中分别放入适量按不同乙腈用量(15、30、50、70 mL)、不同充氮气时间(5、10、15、20 min)、不同聚合温度(50、60、70 ℃)条件下制备的MMIP及MNIP,再分别加入5 mL 0.1 mmol/L联苯三唑醇-乙腈标准溶液,25 ℃恒温振荡24 h,10 000 r/min 离心5 min,静置,取上清液。采用HPLC对聚合物吸附平衡后的浓度进行测定,聚合物对目标物的吸附量用溶液浓度的差值进行计算,平行测定3 次后取其平均值,吸附量计算如式(1)所示。用印迹因子(imprinting factor,IF)评价印迹效果,计算如式(2)所示:

式中:Q为吸附量/(μmol/g);C为联苯三唑醇的初始浓度/(μmol/L);Ce为联苯三唑醇的平衡 浓度/(μmol/L);V为吸附溶液的体积/mL;m为聚合物的质量/mg。

式中:QMIP为印迹聚合物吸附联苯三唑醇 的量/(μmol/g);QNIP为非印迹聚合物吸附联苯三唑醇的量/(μmol/g)。
1.3.4 静态吸附实验
在具塞玻璃管中分别放入适量的MMIP及MNIP,并分别加入5 mL浓度为0~5 µmol/L联苯三唑醇-乙腈标准溶液,25 ℃恒温振荡24 h后,用HPLC检测联苯三唑醇的浓度,并计算平行测定3 次实验结果的平均值,聚合物对目标物的吸附量用溶液浓度的差值计算,以初始溶液浓度C0为横坐标,Q为纵坐标作图,并对其进行Scatchard分析[31]。
1.3.5 MMIP产率的测定
根据制备过程和需要对聚合物进行称量,获得以下质量数据以计算聚合物产率:1)称取干燥的圆底烧瓶(100 mL)质量m1。2)称取加入了反应液(模板分子+ MAA+乙腈)的烧瓶质量m2。3)向已在冰箱中放置过夜的装有反应液的烧瓶中加入EDMA、磁性微球后,称量反应体系的质量m3。4)加入AIBN,超声除氧气,60 ℃水浴反应24 h,此过程宜避光进行。5)待反应完成后,自然放置至室温,称量其质量m4。6)将反应体系中的固体聚合物与液体分离开,待其自然晾干后,称量装有聚合物的烧瓶质量m5。7)向上述装有聚合物的烧瓶中加入适量的乙腈,浸洗2 次后,待其自然晾干,称量装有聚合物的烧瓶质量m6。8)用甲醇-乙酸(9∶1,V/V)对聚合物进行超声,使其分散,然后洗脱去除模板分子,再用甲醇洗去残留在聚合物中的乙酸,最后在真空干燥箱内(60 ℃、0.04 MPa)进行干燥。称量装有聚合物的烧瓶质量m7(本实验采用的计算方法中聚合物的质量不包括模板分子的质量,因此需要洗脱掉聚合物中的模板分子再对其进行称量),聚合物产率计算 如式(3)所示:
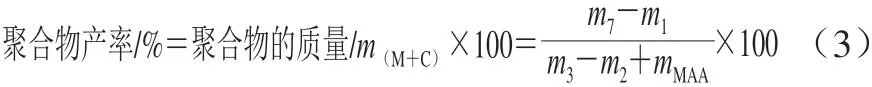
1.3.6 聚合物的表征
1.3.6.1 傅里叶变换红外光谱的测定
表征样品与溴化钾混合后进行研磨、压制,于400~4 000 cm-1范围内对Fe3O4、Fe3O4@SiO2、MMIP和MNIP进行扫描,得到所对应的红外光谱,扫描扣除CO2干扰。
1.3.6.2 透射电子显微镜的测定
将少量Fe3O4、MMIP放入离心管中,用无水乙醇对其溶解,超声使其分散均匀,分散后取一小滴放到铜网上,用红外灯照射使其干燥,然后用透射电子显微镜进行测定。
1.3.6.3 磁响应性能的测定
称取少量MMIP置于10 mL离心管中,加入5 mL乙腈溶液,超声10 min混合均匀。在离心管侧面放置磁铁,并连续拍照,记录聚合物的磁分离过程。
1.3.7 样品测定
1.3.7.1 样品的预处理
分别称取样品(小米、小麦、白菜、梨、苹果等)10.000 g,磨碎或切碎,果蔬样品中加入无水硫酸钠5.000 g,除去过多的水分(谷物中不加),用20 mL 乙腈溶解,进行提取(分2 次,每次10 mL),搅匀,超声20 min,1 000 r/min离心10 min,吸取上清液1 mL经MMIP萃取,用20 mL水淋洗,再用10 mL甲醇-乙酸(9∶1,V/V)洗脱,收集洗脱液进行检测[32]。
1.3.7.2 MMIP对模板分子的磁萃取分离与测定
称量10 mg MMIP置于10 mL具塞三角瓶中,加入5.00 mL试样液,室温条件下,150 r/min振荡5 h,磁分离收集平衡液。然后用2 mL乙腈分2 次洗去聚合物微球表面非特异性吸附的干扰物质,磁分离弃去洗涤液。再用2 mL甲醇-乙酸(9∶1,V/V)分2 次将吸附在磁性聚合物上的目标化合物洗脱下来,磁分离收集洗脱液。然后,对洗脱液进行蒸发、干燥,用1.00 mL甲醇对其进行复溶,用0.45 μm微孔滤膜过滤,待HPLC检测。
1.3.7.3 HPLC测定条件
1.4 数据统计及图表绘制
采用Origin 8.0和Excel 2007对实验数据进行作图及统计分析,每组实验重复测量3 次,数据均为平均值。
2 结果与分析
2.1 聚合条件对聚合物吸附性能的影响

表1 聚合条件对聚合物吸附性能的影响Table 1 Effect of different polymerization conditions on the adsorption properties of polymers
固定联苯三唑醇、功能单体和交联剂的条件不变,通过考察乙腈用量、充氮气时间、聚合温度研究其对聚合物吸附性的影响。从表1可见,乙腈50 mL、充氮气15 min、聚合温度60 ℃时吸附量大且印迹效果好。因为,当乙腈用量为15 mL时,聚合反应与本体聚合效果相当,即生成的聚合物为块状;随着乙腈用量的增加,乙腈不仅起到溶解作用,而且也有致孔作用,导致聚合物会更加松散,当乙腈用量为50 mL时能形成较多聚合物,但当乙腈用量增加到70 mL时,聚合物不易形成,因此,实验选择50 mL为最佳乙腈用量。实验研究了充氮气时间,当冲入氮气5 min时,产生的聚合物很少,这是由于充入的氮气量少不能将氧气完全赶出;充氮气20 min时产生的聚合物明显少于充氮气15 min,这是因为充气时间过长会导致致孔剂挥发;而充氮气10 min时生成聚合物所需时间过长,只有在充氮气15 min条件下,反应8 h左右聚合物基本全部生成。实验还研究了聚合温度对聚合物的影响,随着温度的升高,聚合速率加快,70 ℃时,在6 h能生成大量的MMIP;60 ℃时,8 h左右出现大量MMIP;50 ℃时,反应24 h以后,能够生成较多的MMIP;实验还尝试在45 ℃聚合,反应48 h以后,反应液仍为澄清,无聚合物生成。结果表明,聚合温度过高(如70 ℃)时,体系容易发生爆聚,导致聚合物结构不均一;而低温制备的聚合物其分子识别性好,但是如果温度过低,就不能达到引发剂AIBN进行热分解的温度,不能发生聚合反应,因此实验中选用60 ℃作为最佳反应温度。
2.2 聚合物的吸附性能
2.2.1 静态吸附等温线

图2 MMIP和MNIP对联苯三唑醇的静态吸附等温线Fig. 2 Static adsorption isotherms of MMIP and MNIP toward bitertanol
以初始质量浓度C0为横坐标、吸附量Q为纵坐标作图,得到MMIP和MNIP的静态吸附等温线,如图2所示。可以看出,MMIP比MNIP对模板分子有更好的吸附性能,在相同的浓度下,MMIP的吸附量远大于MNIP,这是由于在MMIP的合成过程中其内部形成的空间结构能够与模板分子吻合,形成印迹空穴,它可以“记住”模板分子,可以特异吸附模板分子。而MNIP中不存在模板分子,也不能形成印迹空穴,因此MNIP对模板分子无特异吸附性。
2.2.2 Scatchard分析
使用Scatchard模型对聚合物的吸附性能进行评价,如式(4)所示:

式中:Qmax为结合位点的最大表观量/(μmol/g);Q为MMIP吸附模板分子的量/(μmol/g);C0为模板分子的初始质量浓度/(μmol/L);Kd为结合位点的平衡解离常数。
1.4 统计学分析 HaploView 4.2软件进行SNPs的 Hardy-Weinberg平衡、连锁不平衡水平(LD)分析。SPSS 19.0统计软件分析入组人群与其他地区人群的差异,χ2检验分析基因型及等位基因频率的统计学意义,P<0.05为差异有统计学意义。
以Q为横坐标,Q/C0为纵坐标绘制Scatchard曲线,如图3所示。

图3 MIP吸附性的Scatchard曲线Fig. 3 Scatchard curve of MIP adsorption
图3 表明,Q/C0与Q不存在单一的线性关系,表示MMIP中的结合位点不是等价的,说明MMIP吸附目标物质时,作用位点非均一,其具有2 类结合位点,分别为特定孔穴引起的高结合位点和一些非共价键引起的低结合位点,每种结合形式有独特的作用性质。由图3可看到2 条直线,故可推断它们之间有2 种结合位点,说明联苯三唑醇与MAA在形成聚合物时有2 种结合形式,并且在洗脱模板分子后形成2 种结合性质不同的结合位点。通过作图拟合,得到2 个线性方程:第1类吸附位点的方程为y=62.051-0.337x,Kd1=2.97 µmol/L,Qmax1= 184.29 µmol/g;第2类吸附位点的方程为y=0.070 7x+26.089,Kd2=14.14 µmol/L,Qmax2=368.90 µmol/g。
2.3 聚合物产率分析
在固定其他反应条件不变的前提下考察聚合物产率随聚合时间的变化关系,结果显示当聚合时间为24 h时,聚合物产率最高;当聚合时间超过24 h后,聚合物产率增加幅度趋于平稳,因此确定最佳聚合时间为24 h。在此条件下改变引发剂使用量考察引发剂用量对聚合物产率的影响。结果表明,随着引发剂用量的增加,功能单体和交联剂聚合的愈发充分,聚合物产率逐渐增加;当引发剂用量超过40 mg时,聚合物产率增加的速度趋于平缓,这是因为此时分散体系中可以利用的功能单体和交联剂都消耗殆尽,化学反应接近终点,无法形成更多的聚合物。综上所述聚合时间为24 h且引发剂用量为40 mg时聚合物产率最高,最高产率为76.4%。
2.4 MMIP的表征
2.4.1 红外光谱分析
由图4a可见,572 cm-1为Fe—O的特征吸收峰,说明已制备了Fe3O4粒子。图4b中1 097、952、563 cm-1处分别为Si—O—Si的反对称伸缩振动峰,Si—O—H的弯曲振动吸收峰和Fe—O的特征峰,794 cm-1和468 cm-1处分别为Si—O的伸缩和弯曲振动峰,这些特征峰的增加说明SiO2已成功包裹在Fe3O4粒子上。图4c为MMIP的红外谱图,3 440、1 094、948、578 cm-1处分别为硅胶表面的O—H基团的伸缩振动峰、Si—O—Si反对称伸缩振动峰、Si—O—H的弯曲振动吸收峰和Fe—O的特征峰,803 cm-1和463 cm-1处分别为Si—O的伸缩和弯曲振动峰,除此之外,2 990、1 736 cm-1处为饱和C—H的伸缩振动吸收峰和C=O伸缩振动吸收峰,1 631 cm-1处对应的C=C伸缩振动吸收峰很小,说明交联剂与功能单体大部分进行了交联聚合反应,这些特征峰的增加说明在包裹了SiO2的Fe3O4表面有聚合物生成。图4d为MNIP的红外谱图,它的吸收峰位置与图4c的吸收峰位置基本一致,但1 736 cm-1处C=O伸缩振动吸收峰相对于1 093 cm-1处Si—O—Si反对称伸缩振动峰,相对峰值明显减小,说明在磁性微球表面发生聚合的MNIP较少。结果表明,功能单体MAA与模板分子联苯三唑醇在磁性微球表面预聚合形成复合物时有利于聚合物的聚合。MNIP的吸收峰位置与MMIP基本一致,证明MMIP与MNIP 2 种交联共聚物骨架结构的化学组成相同。结果表明,在包覆SiO2的磁性Fe3O4载体表面有印迹聚合反应发生。
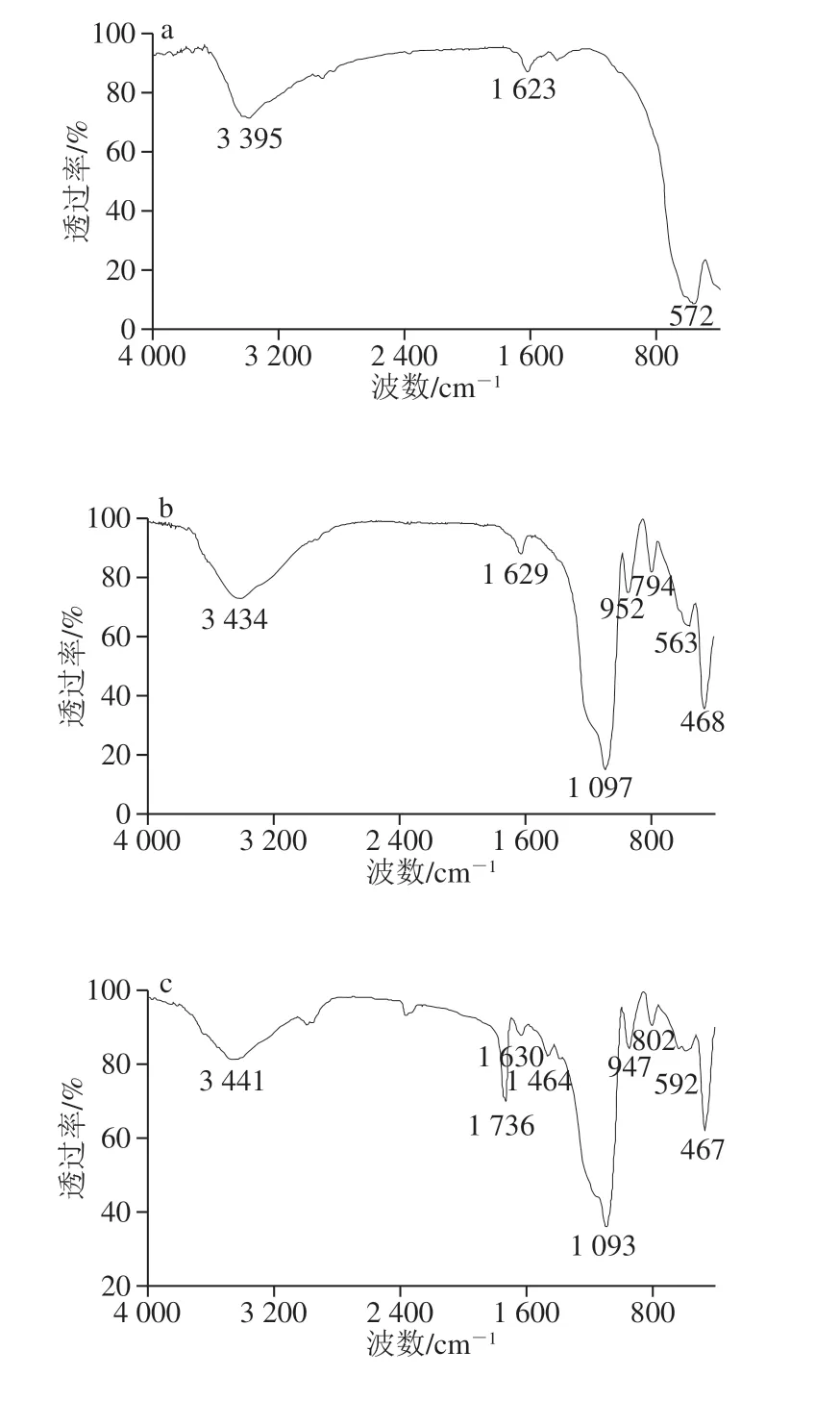

图4 Fe3O4(a)、Fe3O4@SiO2(b)、MMIP(c)和 MNIP(d)的红外光谱图Fig. 4 IR spectra of Fe3O4 (a), Fe3O4@SiO2 (b), MMIP (c) and MNIP (d)
由联苯三唑醇的分子结构分析可知,联苯三唑醇分子中含有三唑环,存在着带有孤对电子的N原子,电负性较大,可与羧基中的H形成氢键;联苯三唑醇分子中还含有羟基,可与MAA中羰基上的O形成氢键;此外联苯三唑醇分子还显示一定的碱性,可以和功能单体MAA的羧基产生离子作用;由此可推测印迹分子联苯三唑醇与功能单体MAA的作用形式,如图5所示。
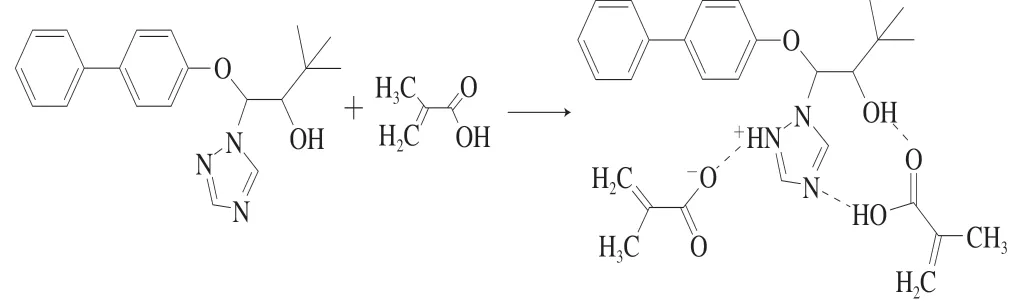
图5 联苯三唑醇与功能单体作用形式Fig. 5 Action form between bitertanol and functional monomer
2.4.2 透射电子显微镜分析
通过透射电子显微镜表征Fe3O4和MMIP形貌,由图6A可知,Fe3O4为大小约10 nm的球形粒子,且有一定的团聚,说明制备所得Fe3O4为纳米级粒子。由图6B可知,MMIP粒径约为100 nm,其粒径较Fe3O4粒子大很多,说明在Fe3O4表面确实形成聚合物层。2.4.3 磁响应性能分析

图6 Fe3O4(A)和MMIP(B)的透射电子显微镜图Fig. 6 Transmission electron micrographs of Fe3O4 (A) and MMIP (B)
为了考察印迹聚合物的磁响应性能,将其溶于乙腈溶剂中,超声使其均匀分散,并将其置于磁铁的一侧,每隔1 s拍照一次,记录其在外加磁场下与溶液的分离过程,拍摄的照片如图7A所示。实验发现,MMIP在磁场作用下,能迅速聚集在与磁铁右端接触的管壁上,当与磁铁接触10 s时MMIP即完全聚集在管壁上,溶液澄清。当撤走外加磁场后,聚合物粒子很快会重新分散在乙腈溶剂中,得到稳定的磁流体,如图7B所示。结果表明,在乙腈溶剂中,MMIP具有良好的磁响应和再分散性能。

图7 在外磁场作用下印迹聚合物聚集过程(A)和 分散性能(B)的照片Fig. 7 Photographs showing the aggregation process of imprinted polymers (A) under external magnetic field and dispersion performance (B)
2.5 线性范围与检出限
实验将4 种三唑类农药储备液(100 μg/mL)分别配制成不同质量浓度(0.01~100 μg/mL)的标准溶液,经测定后,以待测物质量浓度为横坐标,峰面积为纵坐标绘制标准曲线,结果如表2所示。联苯三唑醇线性范围为0.06~100 μg/mL,三唑酮、戊唑醇和烯唑醇的线性范围为0.03~100 μg/mL,线性关系好(r≥0.999 6),检出限在0.01~0.02 μg/mL之间。

表2 4 种三唑类农药线性关系及检出限Table 2 Linear relationships and detection limits for four triazole pesticides
2.6 联苯三唑醇MMIP对样品的净化和吸附能力
以联苯三唑醇MMIP作为固相萃取材料,样品按1.3.7.1节方法处理,在1.3.7.3节HPLC测定条件下进行测定。结果显示,本实验所测的多种样品中均未检出上述4 种三唑类农药。代表性样品的色谱分离图见图8、9。
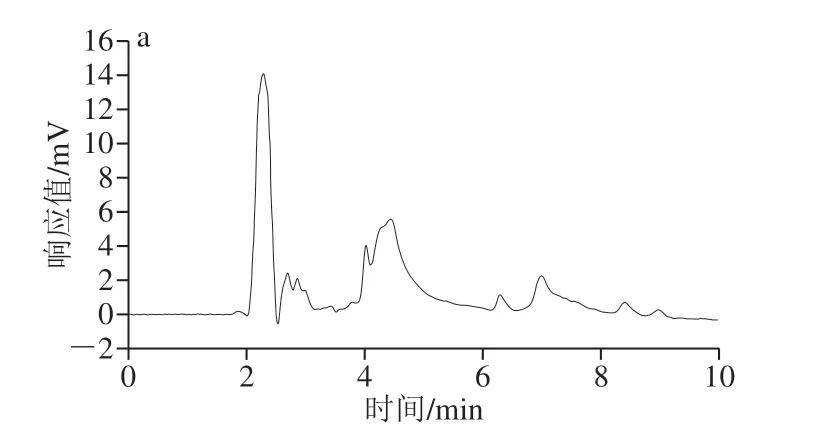
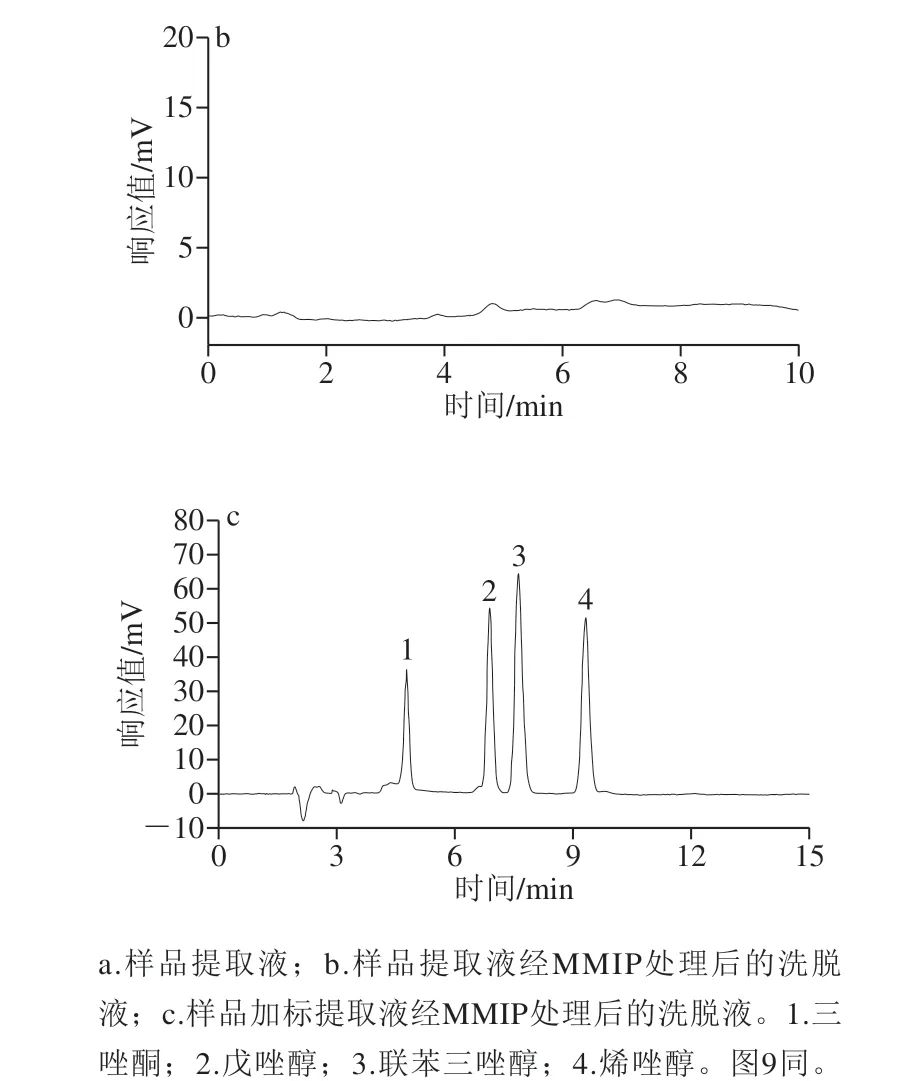
图8 分子印迹固相萃取小米样品吸附净化效果谱图Fig. 8 Chromatograms of four triazole pesticide residues extracted from mille by molecularly imprinted solid phase extraction

图9 分子印迹固相萃取梨样品吸附净化效果谱图Fig. 9 Chromatograms of four triazole pesticide residues purified from pear fruit by molecularly imprinted solid phase extraction
由图8a、b与图9a、b可知,聚合物不能吸附杂质,可以很好地净化样品。由图8c和图9c可知,磁性分子印迹萃取材料只能吸附模板分子和它的结构类似物,这充分表明了它可以很好地分离和富集复杂基质中的目标物质。
2.7 回收率和精密度实验结果
采用本方法对小米、小麦、白菜、梨、苹果等多种样品在添加量为0.1 μg/mL和10 μg/mL条件下进行测定,其结果见表3。
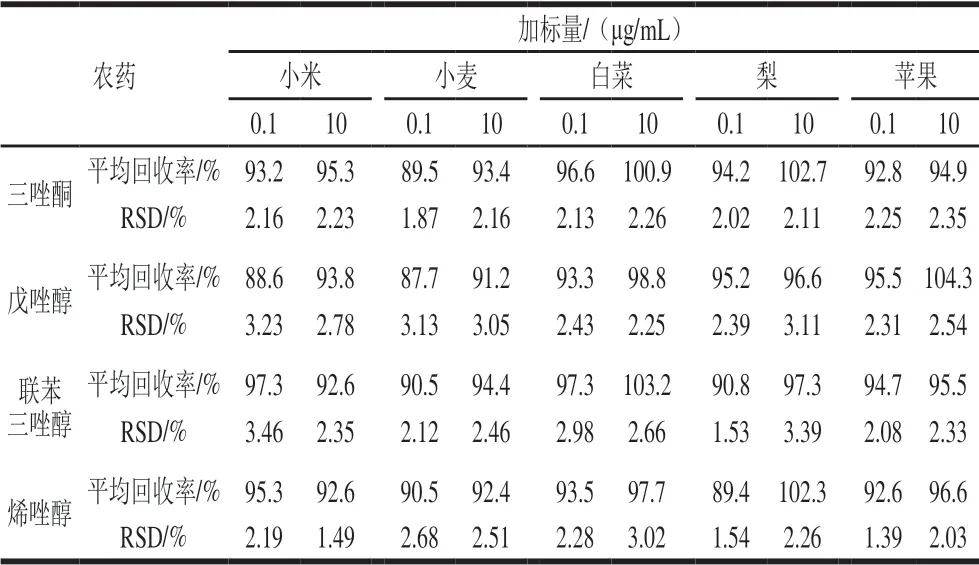
表3 回收率和精密度实验结果(n=5)Table 3 Recovery and precision of the proposed method (n= 5)
由表3 可知, 4 种农药的平均回收率在87.7%~104.3%之间,相对标准偏差(relative standard deviation,RSD)为1.39%~3.46%(n=5)。该检测方法对三唑类农药的回收率高、精密度好。将实验制备的印迹传感器与其他文献方法检测实际样品进行比较,如表4所示,该方法样品前处理简便,且检出限更低。

表4 该方法与其他测定联苯三唑醇的方法比较Table 4 Comparison of this method with other methods for the determination of bitertanol
3 结 论
以Fe3O4@SiO2为磁性载体,联苯三唑醇为模板分子,通过表面印迹的方法,合成了具有核壳结构的聚合物微球,并优化了聚合工艺,当致孔剂乙腈用量50 mL、聚合温度60 ℃、充氮气时间15 min时IF最高,特异性吸附能力最好,且聚合物产率为76.4%。静态吸附结果表明,MMIP有很好的吸附性;由Scatchard分析可知,联苯三唑醇和MAA有2 种结合位点,Kd1=2.97 µmol/L、Kd2=14.14 µmol/L。通过红外光谱、透射电子显微镜及磁响应对聚合物进行表征,结果表明,MMIP聚合成功印迹聚合物粒径约为100 nm,且聚合物磁响应性能和再分散性能较好。以最优条件下制备的聚合物为固相萃取材料,对多种样品进行萃取净化,并采用HPLC法进行测定。结果为样品加标回收率为87.7%~104.3%,RSD为1.39%~3.46%(n=5)。该方法简单快速、选择性强,有利于促进样品的快速前处理实现。
