热敏型可修复分子印迹固相微萃取纤维的制备及其应用
2020-12-29郭跃龙纪顺利
郭跃龙,吴 丹,郑 枫,纪顺利*
(1江苏省省级机关医院药剂科,南京210024;2中国药科大学药物分析教研室,南京210009)
大环内酯类抗生素(macrolides antibiotics,MACs)是一类分子结构中具有12~16 碳内酯环的药物总称,主要用于治疗需氧革兰阳性球菌和革兰阴性球菌感染所引发的疾病[1],不良反应包括消化道症状、肝毒性、心脏毒性等。日常生产生活中,抗生素常被添加到饲料中促进动物生长,部分没有代谢的抗生素最终在人体内蓄积,进而影响人体健康。许多国家的监管机构都制定了食品基质中MACs 的最大残留限定(MRLs),因此,开发快速有效的食品基质中MACs 的定量检测技术非常必要。
MACs 结构中缺乏发色团,紫外响应弱,不能使用紫外检测器对含量低的样品直接定量检测[2];此外,食品基质复杂,可能会干扰目标物的检测。为富集目标物以及屏蔽基质干扰,需要开发合适的样品前处理方法。常用的样品前处理方法有固相萃取[3-4]、基质分散固相萃取[5]、QuEChERS[6]和固相微萃取(SPME)等。其中,SPME 是一种集采样、萃取和浓缩于一体的方法,适用于痕量化合物的提取和净化。SPME 的核心在于萃取头,传统萃取头以非特异性吸附为主,选择性、热稳定性和化学稳定性差,因此制备可重复利用、具有良好选择性和稳定性的新型涂层材料是非常值得关注的。
分子印迹聚合物(molecular imprinting polymer,MIPs)是一种稳定性高、具有特异识别功能的新材料,可通过模板分子和功能单体与交联剂聚合而获得。聚合后移除模板分子,会留下特定的空穴,可以根据大小、形状和功能基团选择性地“锁定”并富集目标化合物,适用于各种复杂样品的前处理。近年来,报道了多种分子印迹材料对食品样品中的红霉素或替米考星进行选择性富集的材料,诸如磁性分子印迹聚合物[5]、固相萃取[7]和固相微萃取(SPME)[8]等。但目前尚无有关热敏型、可修复的分子印迹固相微萃取纤维(MIPfibers)制备方法的报道。本研究以螺旋霉素为模板分子,N-异丙基丙烯酰胺和甲基丙烯酸为功能单体,乙二醇甲基丙烯酸酯为交联剂,硅烷化的石英毛细管为载体合成了具有热敏型、可修复的、能够特异性识别MACs 的MIP-fibers,并用其同时检测食品基质中多种MACs。
1 材 料
1.1 试 剂
替米考星(TILM)、螺旋霉素(SPI)、交沙霉素(JOS)、泰乐菌素(TYL)(德国Dr.Ehrenstorfer 公司);阿奇霉素(AZI)、克拉霉素(CLA)、罗红霉素(ROX)(中国食品药品检定研究院);磺胺嘧啶(美国Alfa Aesar 公司);盐酸土霉素、恩诺沙星、偶氮二异丁腈(美国Sigma-Aldrich 公司);3-(甲基丙烯酰氧)丙基三甲氧基硅烷(美国Acros 公司);N-异丙基丙烯酰胺(上海麦克林生化科技有限公司);甲基丙烯酸(MAA)、4-乙烯基吡啶、丙烯酰胺、乙二醇甲基丙烯酸酯(中国上海阿拉丁试剂有限公司);甲醇、乙腈均为色谱纯;其余试剂均为分析纯。实验用水由Milli-R04净化系统(美国Millipore公司)制得。
1.2 仪 器
Alliance 2695 高效液相色谱仪(美国Waters 公司);FESEM NOVA NanoSEM450 扫描电子显微镜(美国FEI 公司);TriStarII3020TM比表面积和孔体积分析仪(美国Micromeritic公司)。
2 方 法
2.1 溶液配制
准确称取适量7 种MACs 标准品,用甲醇-水(20∶80)溶解并定容,配制成1 mg/mL 的标准储备液,于4 ℃冰箱中保存,在使用前用20 mmol/L K2HPO4溶液(pH 7.4)稀释定容制成混合标准工作液。恩诺沙星、磺胺嘧啶和盐酸土霉素除用甲醇溶解外,其余处理方法均与MACs相同。
2.2 MIP-fibers的制备
截取长约4 cm 的石英毛细管(50 μm × 365 μm),火烧去除一半长度的外壁保护层。将毛细管依次浸泡在1 mol/L NaOH 和0.1 mol/L HCl 溶液中、去离子水洗至中性后氮气吹干。毛细管硅烷化过程参照文献[9]中方法:将上述毛细管浸泡在3-(甲基丙烯酰氧)丙基三甲氧基硅烷-丙酮(1∶9)溶液中2 h后,100 ℃下干燥2 h。
热敏型MIP-fiber 参照文献[10]中的最佳实验条件制得:称取SPI 0.1 mmol、甲基丙烯酸0.42 mmol、N-异丙基丙烯酰胺0.4 mmol 溶于二甲基亚砜-氯仿(1∶2)溶液3 mL 中,混合均匀后加入乙二醇甲基丙烯酸酯2 mmol和偶氮二异丁腈10 mg,即得预聚合物溶液。将毛细管硅烷化的一端插入2 cm 长的玻璃管(0.9~1.1 mm)后,放进4 mL 的离心管中,加入制备好的预聚合物溶液,60 ℃水浴条件下反应24 h。待反应结束后,将毛细管从玻璃管中推出,即得MIP-fiber。将MIP-fiber 浸入乙酸-甲醇(1∶4)中,涡旋5 min 后去除模板分子后保存在甲醇中,待后续实验使用。
非印迹固相微萃取纤维(NIP-fibers)的制备过程中不加入SPI,其余合成步骤与MIP-fibers相同。
2.3 色谱条件
MACs 是一种碱性抗生素,用普通的C18色谱柱通常会出现拖尾现象,因此选用表面带正电荷的Acchrom XCharger-C18型反相色谱柱(150 mm ×4.6 mm,5 μm)来解决该问题。流动相:乙腈(A),0.02% 磷酸水溶液(B);梯度洗脱程序:0~9 min,A 相从5% 到44%;柱温:30 ℃;流速:1 mL/min;进样体积:10 μL;检测波长:通道1(283 nm),通道2(232 nm),如图1。

Figure 1 Separation of 4 macrolides antibiotics(MACs)A:Spiramycin (SPI);B:Josamycin (JOS);C:Tilmicosin (TILM);D:Tylosin(TYL)
2.4 样品预处理
用4 种不同来源的蜂蜜样品来评价MIP-fiber的富集效果,选择洋槐花蜂蜜作为空白样品(经检测该蜂蜜不含目标抗生素)。样品预处理方法如下:称取空白样品2 g(精确至0.01 g),置于10 mL离心管中,加入20 mmol/L K2HPO4(pH 7.4)缓冲溶液5 mL,充分涡旋混合30 s 后,以4 500 r/min 离心10 min,收集上清液,作为SPME上样液。
2.5 净 化
净化流程如图2 所示:将制备好的萃取纤维(MIP-fiber 或NIP-fiber)浸没在样品溶液中,500 r/min 下搅拌60 min。然后将纤维取出,用乙腈-水(1∶4)1.5 mL 淋洗,最后采用涡旋法45 ℃条件下洗脱2 min,洗脱液为乙酸-乙腈(1∶4)1.5 mL。洗脱液氮气吹干后,用甲醇-水(1∶4)200 μL 复溶,进行紫外检测。
3 结果与讨论
3.1 去除模板分子
去除模板分子是制备分子印迹材料中最关键的一步,但是通常存在耗时较长和难去除彻底的问题,为此,以甲醇-乙酸(8∶1)作为洗脱液,测试了两种不同的去模板方法。(1)振荡法:将3~5 根MIP-fiber放进加有洗脱液的离心管中,以200 r/min的条件振荡24 h,每8 h 更换一次洗脱液。该方法可有效去除SPI,但所需时间长,在振荡过程中纤维之间的摩擦容易造成MIP 材料的损失。(2)涡旋法:采用相同的洗脱液涡旋洗脱5 min,重复该洗脱过程直至无模板被检出。采用该方法使得萃取纤维在洗脱液中高速旋转,可快速去除模板分子且不会损失涂层材料,所以选择涡旋法去除模板分子。
3.2 MIP-fibers制备条件的优化
分别以MAA、丙烯酰胺(AM)、4-乙烯基吡啶(4-VP)为功能单体,改变模板与功能单体之间的物质的量比制备5 种MIP-fibers,将制得的MIPfibers 分别放入20 μg/mL SPI 溶液5.0 mL 中,45 ℃水浴条件下萃取120 min 后使用高效液相色谱法测定溶液中SPI 的浓度,结果如表1 所示,可以看出MAA 作为单体时的吸附能力最强;模板分子与MAA 的物质的量比为1∶4.2 时达到最大吸附量82.5%。
3.3 表 征
图3-A 为MIP 反应前后毛细管的对比照片,经MIP 反应后,毛细管外可见均匀的印迹聚合物涂层。MIP-fiber 和NIP-fiber 的扫描电子显微镜照片(SEM)如图3 所示,相较于NIP(图3-C),MIP(图3-B)具有更多的孔隙结构和更高的交联度,因此具有更大的表面积,从而有较高的吸附容量。MIPfiber干燥状态下的SEM如图3-D所示,可以观察到许多断层,说明聚合物材料在干燥环境和溶液中的柔韧性不同,这与实际应用过程中发现的现象是一致的。有趣的是将干燥后断裂的MIP-fibers浸泡在甲醇溶液中一段时间后,纤维上的断层消失,恢复到刚制备出来的形态。因此,判断MIPfibers有自修复功能。

Table 1 Optimization of functional monomer
采用氮气吸附孔隙度测定法测定MIP/NIP 材料的比表面积和孔容,MIP、NIP的比表面积分别为130.928、1.761 m2/g,孔 容 分 别 为0.294、0.017 cm3/g。与SEM 检测所得到的结果相同,MIP 材料的比表面积远远大于MIP 材料,这充分说明所制备的MIP-fiber非常适合于样品前处理。

Figure 3 Photographs and SEM images of molecular imprinting polymer(MIP)-fiber and non-imprinting polymers (NIP)-fiberA:Fibers after molecular imprinting (1) and before molecular imprinting(2);B:Morphology of MIP-fiber;C:Morphology of NIP-fiber;D:SEM image of MIP-fiber in dry condition
3.4 静态吸附和吸附动力学实验
吸附容量和速率是影响固相微萃取吸附剂性能的重要因素。
3.4.1 静态吸附实验 分别将一根MIP-fiber/NIP-fiber 放入装有30~900 μg/mL SPI 溶液5 mL的离心管中,密封后45 ℃水浴条件下搅拌24 h。HPLC-UV 测定溶液中SPI 的残留量,并根据公式(1)计算MIP-fiber/NIP-fiber吸附容量。

其中:c(mg/mL)、ct(mg/mL)分别代表SPI 初始、结束时的质量浓度;V(mL)代表样品体积;m(g)代表涂层质量。
得到结果如图4-A 所示,在SPI 浓度较低时二者差异很小,随着初始溶液中SPI 浓度的增加,二者的吸附量都有所提高,但是MIP-fiber 比NIPfiber 提高得更明显,表明MIP-fiber 的吸附性能优于NIP-fiber。
3.4.2 吸附动力学实验 将一根MIP-fiber/NIPfiber 放入100 μg/mL SPI 溶液25.0 mL 中,密封后45 ℃水浴条件下搅拌240 min,分别在1、5、10、15、20、25、60、90和240 min时进行取样,检测SPI的浓度。得到结果如图4-B 所示,MIP-fiber 在60 min 内达到吸附平衡,NIP-fiber 在240 min 后仍未达到吸附平衡,表明MIP-fiber具有更稳定的吸附性能。

Figure 4 Adsorption isotherms (A) and adsorption kinetic (B) curves of SPI on MIP-fibers and NIP-fibers (n=3)
3.5 萃取条件的考察
固相萃取通常包括萃取、淋洗和洗脱3 步,为了得到最佳富集效果,本研究对几个可能影响萃取效率的参数进行了优化。除特殊说明外,其他样品处理及固相微萃取条件均采用“2.4”和“2.5” 项中方法,并重复操作3次。
3.5.1 温度的影响 由于N-异丙基丙烯酰胺和MAA的加入,使得MIP-fiber同时具有pH和温度响应的立体选择性。其根本原因为温度会改变MIP聚合物内部孔道的大小,使得MIP-fiber 在较低温度下膨胀,在较高温度下收缩,从而与目标化合物的匹配程度不同。为此研究了MIP-fiber 在不同温度下对4种MACs的吸附情况,结果如图5-A 所示,验证了以上结论,同时在45 ℃时MIP-fiber 与模板分子SPI的匹配程度最高,因此在后续试验中采取45 ℃作为萃取温度。
3.5.2 pH 的影响 蜂蜜溶液的pH 在3 附近,因此本实验选择pH 为3~10 的K2HPO4缓冲盐溶液(20 mmol/L)对蜂蜜进行稀释后进行萃取,结果如图5-B 所示。萃取体系pH 逐渐升高,4 种MACs 的萃取效率均呈现“先升后降”趋势,并在pH 7.4 时达到最高,因此,后续实验中,使用pH 为7.4 的K2HPO4溶作为蜂蜜的稀释溶液。
3.5.3 淋洗条件 良好的淋洗溶液可以在不影响目标物回收率的前提下洗去非特异性保留的物质,从而达到屏蔽基质及其他杂质干扰的目的。本研究使用4 种不同的淋洗溶液,结果如图5-C 所示,4种淋洗溶液均会对目标物造成一定的损失,但去离子水和乙腈淋洗后损失较少,分别损失0.15%和4.46%。因此进一步研究了乙腈与水不同比例时对MACs 回收率的影响,发现乙腈-水(1∶4)溶液能有效消除基质干扰,且无MACs的损失。最终选择乙腈-水(1∶4)作为后续实验的淋洗溶剂。
3.5.4 洗脱条件 为了尽量将目标物完全洗脱,从而得到满意的回收率,对洗脱溶液的种类及比例进行了考察。选取甲醇、丙酮、乙醇和乙腈4 种洗脱溶液,结果发现使用乙腈溶液进行洗脱效果最好;进一步考察了乙腈中乙酸比例的影响,洗脱液中待测物峰面积如图5-D 所示,乙酸含量在0%~20% 之间时,增加乙酸用量可提高洗脱效果,并在20%时达到最大值,因此选择乙酸-乙腈(1∶4)作为后续实验的洗脱溶液。

Figure 5 Effect of temperature(A), pH(B),eluent(C)and percentage of acetic acid(D)on solid-phase microextraction(SPME)efficiency (n=3)
3.6 富集倍数测定
固相微萃取的富集原理是目标物在样品基质和吸附层之间达到分配平衡,因此溶液中目标物的质量浓度会影响萃取效率。本研究控制SPI 总量为10 μg,配制体积分别为5、20、200 mL 3 种不同质量浓度的溶液,探究在不同质量浓度下MIPfiber/NIP-fiber 对目标物的富集效果,目标分子的富集因子EF通过公式(2)计算得到:

其中:c0和c1分别为标准溶液直接进样的质量浓度及富集前样品溶液的质量浓度;A0和A1分别为直接进样得到的峰面积及富集后的峰面积大小。
结果见图6,可以看出MIP-fiber 对MACs 具有较好的富集浓缩能力,尤其是在低浓度条件下,MIP-fiber的富集效果明显优于NIP-fiber。
3.7 选择性实验
为了验证MIP-fiber 可以对MACs 进行特异性选择,选取SPI 和蜂蜜中常被检测到的3 类非大环内酯类抗生素(磺胺嘧啶、恩诺沙星、盐酸土霉素)添加到基质中进行测试。3 类非大环内酯类抗生素的浓度均为5 mg/kg,SPI 的浓度为0.5 mg/kg。实验结果如图7 所示,使用MIP-fiber 富集后,3 种非大环内酯类抗生素峰响应不升反降,说明MIPfiber 对以上干扰物几乎无富集作用,但对SPI 的富集效果非常好。在某种程度上确认了MIP-fiber 上存在特异性识别位点,可以选择性富集目标物、降低杂质效应、排除基质干扰。
3.8 线性范围与检出限

Figure 6 Effects of loading volume on the extraction efficiencyInset:Chromatogram of (A) injection after SPME treatment (5 mL of 2 μg/mL spiramycin standard solution with 2 g honey sample),(B) 10 μg/mL spiramycin standard solution,and (C) direct injection without SPME treatment (5 mL of 2 μg/mL spiramycin standard solution with 2 g honey sample)
为了验证MIP-HPLC-UV 检测方法的可行性,在蜂蜜样品中制备一系列含不同浓度的SPI、JOS、TYL 和TILM 的基质匹配混合标准溶液。如表2 所示,在0.5~50 μg/mL的线性范围内,该方法对4种MACs均表现出良好的线性,相关系数r2高于0.99。在抗生素残留分析中,方法的灵敏度通常表示为检测限(LOD)和定量限(LOQ)。在这项研究中,以特征色谱峰的信噪比S/N ≥3 和S/N ≥10 分别为方法的LOD 和LOQ,4 种MACs 的LOD 范围为23.8~85.4 μg/kg,而LOQ范围为79.2~284.5 μg/kg。
3.9 方法应用
采用MIP-HPLC-UV法对当地购买的蜂蜜样品进行检测,4 种蜂蜜中均未检测到MACs。为了验证检测方法的正确性,本研究在蜂蜜样品中进行1.25、12.5和125 mg/kg 3个浓度的加标试验,结果如表3 所示,目标化合物的回收率(81.8%~119.1%)较好,日间精密度小于13.8%(n=6),日内精密度小于15.5%(n=3),回收率高、重复性好,可用于蜂蜜样品中MACs的测定。

Figure 7 Selectivity of MIP-fibers to non-macrolide antibiotics and macrolide antibioticA:Enrofloxacin;B:Oxytetracycline hydrochloride;C:Sulfadiazine;D:Spiramycin (a is direct injection and b is the eluted fraction after SPME treatment)

Table 2 Linear range,LODs and LOQs of MACs by HPLC-UV (n=3)
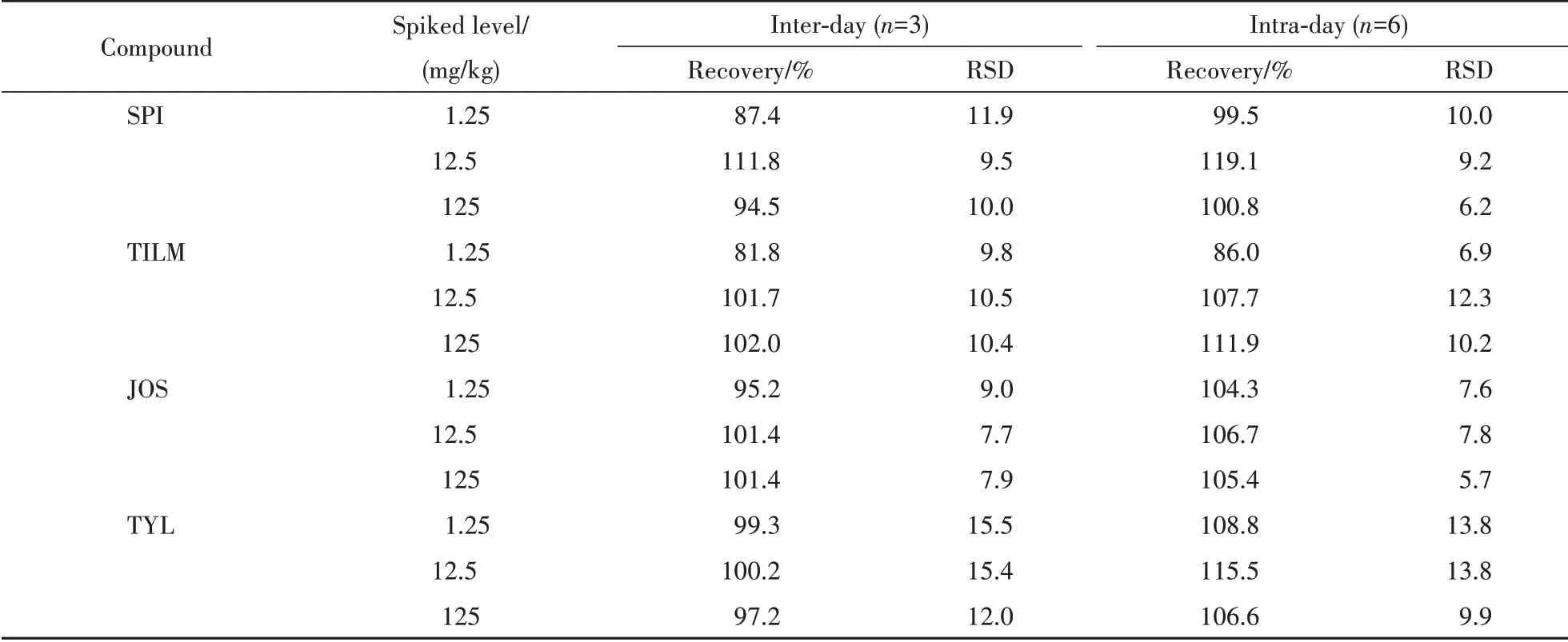
Table 3 Precision and reproducibility of MACs
3.10 液质联用方法验证
上述结果已经证明了MIP-HPLC-UV可以用于检测蜂蜜中MACs的含量以及MACs是否存在超标问题。由于大多数MACs 紫外响应非常弱,上述方法无法实现多种痕量MACs 的同时定量检测。因此本研究进一步发展了基于MIP-fiber的7种MACs的液质联用(HPLC-MS/MS)定量方法,线性范围和检测限如表4所示,更低的检测限和线性范围填补了HPLC-UV的空白,使用者可以根据自身条件、目的及要求对仪器进行选择。
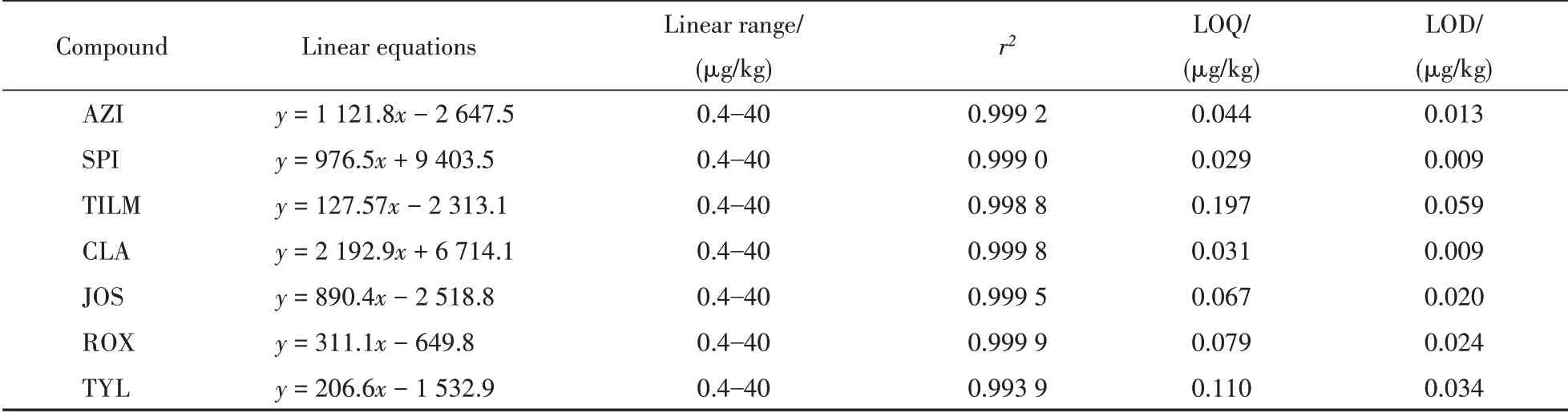
Table 4 Calibration curves,LODs and LOQs of macrolide antibiotics by HPLC-MS/MS (n=3)
采用所开发的MIP-HPLC-MS/MS 法对上述4 种蜂蜜样品的检测结果如表5 所示,仅样品4(洋槐花蜂蜜)中未检测到MACs,其他3种蜂蜜中均含有不同浓度的MACs,但均未超过国家限定标准。
4 结 论
本研究合成了一种具有pH 和温度响应的MIP-fibers,建立了一种快速、高效测定蜂蜜样品中MACs 的方法。该纤维具有一定的自修复功能,对MACs 具有良好的吸附性和选择性,可采用涡旋法快速去除模板和洗脱目标物。样品预处理简单、经济、节省溶剂、节约时间,为分析蜂蜜样品中痕量MACs 的残留提供了一种有效的方法。同时该MIP-fibers 对MACs 具有较好的富集浓缩能力,适用于大体积样品的现场采样及富集浓缩,将其与液相色谱串联质谱联用可实现痕量MACs 的高灵敏检测,后续实验中也将进一步拓展该材料的应用。
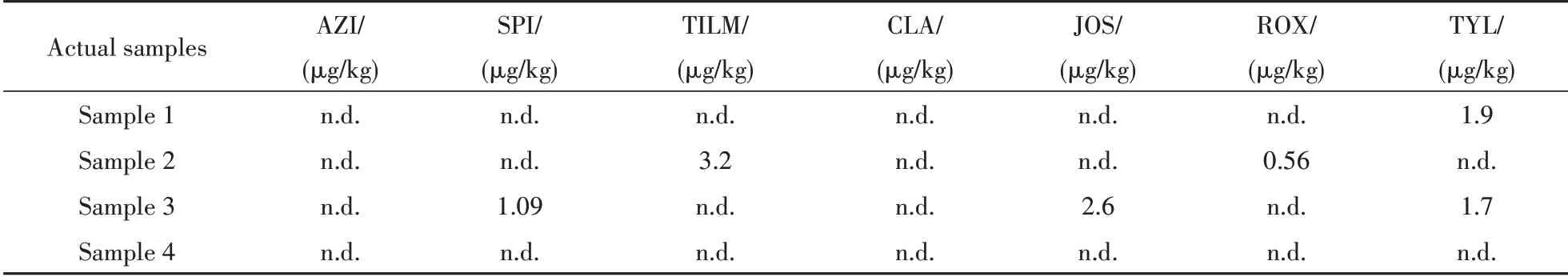
Table 5 Result of the proposed method for honey samples
