ALK抑制剂在NSCLC中的耐药机制及逆转策略的研究进展
2020-12-29冉冬芝甘宗捷
张 迪,冉冬芝,余 瑜,甘宗捷*
(1重庆医科大学药学院,重庆400016;2重庆食品药品检验检测研究院,重庆401121)
间变性淋巴瘤激酶(anaplastic lymphoma kinase,ALK)属于胰岛素受体(insulin receptor,IR)超家族,是一种高度保守的受体型酪氨酸激酶。在成人中,ALK 主要在神经系统,睾丸和小肠中表达并参与调控神经系统的功能和发育[1]。然而,已有研究表明,ALK 的异常表达如基因突变、重排及扩增与多种人类恶性肿瘤如神经母细胞瘤、非小细胞肺癌(non-small cell lung cancer,NSCLC)、卵巢癌等密切相关[2]。例如在NSCLC 中,即检测到约3%~7% 的NSCLC 患者伴有ALK 与棘皮动物微管结合蛋白4(Echinoderm micro tubuleassociated protein-like 4,EML4)的基因融合突变[3],而该融合基因编码形成的EML4-ALK 嵌合蛋白会导致ALK二聚化,从而激活ALK 及其下游RAS/MEK/ERK 以及JAKs/STAT3 等多种信号转导通路,促进细胞的快速增殖与分化,最终导致肿瘤的发生与发展[4]。因此,EML4-ALK 融合基因突变(也称为ALK 阳性突变)已被证明是导致NSCLC 的重要驱动基因突变之一,针对该靶点的ALK 抑制剂的研发一直是当前热点之一[5-6]。
1 ALK抑制剂
近年来,全球已研发出不少针对ALK 的小分子靶向药物,其中,2011 年上市的克唑替尼(crizotinib)是治疗ALK 依赖的NSCLC 的第1 个ALK 抑制剂[7],也是目前NSCLC 伴ALK 阳性患者公认的一线用药,但是由于EML4-ALK 融合蛋白ATP 口袋附近发生的基因突变、基因扩增以及信号通路的旁路激活等因素影响,绝大部分患者在使用克唑替尼约8~12 个月后即出现耐药[8],限制了克唑替尼的临床使用。因此,针对克唑替尼的耐药,又开发了对ALK 激酶专属性和亲和力更强的第2 代ALK 抑制剂色瑞替尼(ceritinib),阿来替尼(alectinib),布格替尼(brigatinib)和第3 代ALK 抑制剂洛拉替尼(lorlatinib)。然而,尽管色瑞替尼、阿来替尼等第2 代ALK 抑制剂能够克服克唑替尼的耐药性,但是长期使用之后也会由于多种原因不可避免的获得性耐药[9]。即使是第3 代ALK 抑制剂洛拉替尼在临床使用过程中也被指出存在有耐药性问题[10]。因此,ALK 抑制剂的耐药问题,严重影响了其在临床上的应用。

2 ALK抑制剂的耐药机制
目前公认的ALK 抑制剂的耐药主要包括原发性耐药和获得性耐药,原发性耐药机制目前尚不明确,初步认为与肿瘤内在因素或患者/药物特异性因素有关[11]。而获得性耐药产生的机制则主要包括ALK依赖型和ALK非依赖型耐药[11-13]。
2.1 激酶结构域的二次基因突变
该机制已被证明与多种激酶类药物耐药的产生密切相关[14],其机制主要是因为激酶结构域内的二次基因突变导致激酶与药物结合区的空间构象发生改变或者增强激酶与ATP 的结合力,从而会影响药物与激酶的结合,致使耐药性的产生。研究表明,约有20%~30% 的克唑替尼耐药患者体内能够检测到二次基因突变,而在第2代ALK抑制剂耐药的NSCLC 患者中,基因突变率更是超过了50%[12],因而二次基因突变已成为NSCLC 肿瘤细胞对第1代、第2代ALK抑制剂耐药的主要原因。
不同ALK 抑制剂诱发的二次突变区不甚相同,比如在克唑替尼耐药的患者中观察到,ALK 激酶域中位于ATP 结合口袋底部的亮氨酸残基L1196,会突变为蛋氨酸(Met),蛋氨酸更长的硫醚侧链会增加空间位阻,从而阻碍了克唑替尼与ALK 激酶的结合,该突变也被称为L1196M“守门员”突变。此外,位于ATP 结合区的甘氨酸残基会点突变为位阻更大的缬氨酸,该突变(G1269A)也是引起克唑替尼耐药的主要突变之一[15]。而对于第2 代ALK 抑制剂(如色瑞替尼、阿来替尼等),突变则主要发生在溶剂前沿区和αC 螺旋的碳末端区,比如G1202R、F1174L 突变等(图1-A)[9]。其中,甘氨酸残基突变为体积更大的精氨酸的G1202R溶剂区突变是第2代ALK抑制剂最常见的基因突变之一,占35%~60%。该突变会引起色瑞替尼和阿来替尼结构中的哌啶环与激酶的结合障碍(图1-B,图1-C)[16],而F1174 突变则会促进ALK活化构象的产生,二者均不利于第2 代抑制剂与ALK的结合,最终导致耐药的产生[17]。
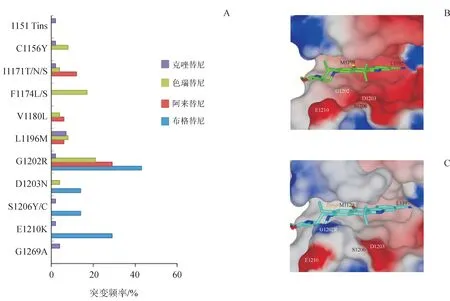
图1 第1代、第2代ALK抑制剂诱发的常见二次基因突变A:各种基因突变的频率[9];B:阿来替尼与ALKwt(PDB:3AOX)的共晶结合模式图;C:阿来替尼与ALK G1202R突变体的共晶结合模式图[16]
2.2 基因扩增与拷贝数增加
ALK 扩增目前仅在少部分克唑替尼耐药的患者发现,而且扩增的发生频率远低于二次突变,因此目前在临床上,基因扩增已不被认为是导致第2代及其之后开发的ALK 抑制剂的主要耐药机制[15,18]。
2.3 旁路信号通路的激活
当ALK 的信号通路被抑制之后,与其密切关联的其他肿瘤蛋白信号通路(如EGFR、KIT、IGF1R 等)会因为反馈机制而补偿性激活[13]。比如,在克唑替尼耐药的细胞株上,就发现了表皮生长 因 子 受 体(epidermal growth factor receptor,EGFR)的异常磷酸化,其下游信号ERK 和Akt 也出现了异常活化,因而肿瘤细胞即使在ALK 通路被阻滞之后,仍然可以继续增生与分化,继而导致了肿瘤对克唑替尼耐药[19]。
2.4 上皮细胞间质转化(epitheliale mesenchymal transition,EMT)
肿瘤细胞表型的改变比如EMT 会使肿瘤细胞获得间质形态并具有迁移和侵袭能力,这也被认为是导致肿瘤转移以及产生耐药的原因之一。研究发现[20],在多种ALK 抑制剂耐药肺癌细胞中可以观察到与EMT 密切相关的间质标记物如波形蛋白(VIM)的表达上调,而上皮标志物如E-钙黏蛋白会下调甚至消失,说明耐药的产生与EMT 有一定关联。
除了以上几种耐药机制,药物转运蛋白P糖蛋白(P-glycoprotein)的过度表达导致的药物外排增加,从而使中枢神经系统(central nervous system,CNS)药物暴露量低引起ALK 抑制剂的耐药也有文献报道[21]。因此,针对ALK 抑制剂的耐药机制,临床上发展了多种策略来克服ALK 抑制剂出现的耐药性问题,并在一定程度上取得了积极的效果。
3 克服ALK抑制剂耐药策略
3.1 与其他药物联用克服耐药
不同抗肿瘤药物的合理联用是目前临床上克服肿瘤耐药性的一种常用手段和方法[22-23]。目前已报道可以通过将ALK 抑制剂与多种其他靶点的抗肿瘤药物联用来克服其存在的耐药性问题。
3.1.1 与Hsp90 抑制剂联用 研究表明,EML4-ALK 融合蛋白是热休克蛋白90(heat shock protein 90,Hsp90)高度敏感的客户蛋白,干扰该分子伴侣蛋白功能可以有效地降低EML-ALK 的表达并抑制ALK 的 激 酶 活 性[24]。 因 此,ALK 抑 制 剂 与Hsp90 抑制剂具有潜在的协同作用。Sang 等[25]就在体内和体外证实了将Hsp90 抑制剂Ganetespib与克唑替尼合用,可以表现出优于克唑替尼的抗肿瘤活性,而且对多种耐药细胞如非小细胞肺癌H3122 克唑替尼耐药细胞株,NB-39-nu 神经母细胞瘤细胞(ALK 扩增型)表现出更强的耐受性和抑制活性。 目前,克唑替尼与Hsp90 抑制剂Onalespib 的联合用药已进入Ⅱ期临床研究(NCT01712217),主要用于NSCLC 的治疗[26]。因此,与Hsp90 抑制剂合用是治疗多种ALK 驱动的恶性肿瘤的潜在有效策略。

3.1.2 与HDAC 抑制剂联用 2018 年,Yun 等[27]的团队从表观遗传学的角度报道了色瑞替尼的获得性耐药产生的作用机制,同时他们也发现,当色瑞替尼与组蛋白去乙酰化酶抑制剂(histone deacetylase inhibitor,HDACi)帕比司他(panobinostat)合用时,可以对AXL 受体的磷酸化及表达产生抑制作用,同时也会下调与EMT 密切相关的神经-钙黏素(CDH2)、波形蛋白的表达,通过以上协同作用,增强了色瑞替尼对耐药细胞的敏感性及抑制活性,使其对H3122 耐药细胞株及其体内小鼠移植瘤产生了强效的抑制作用,最终达到逆转耐药的目的。此外,Fukada 等[28]也开展了类似的研究工作,他们也发现HDAC 抑制剂Quisinostat 可以使克唑替尼对耐药细胞重新敏感。Shen 等[29]发现HDAC8 亚型抑制剂PCI-34051 与克唑替尼合用可以有效杀死ALK 野生型、ALK 扩增型、ALKF1174L突变型等神经母细胞瘤细胞株。

3.1.3 与CDK 抑制剂联用 细胞周期蛋白依赖性激酶(cyclin dependent kinase,CDK)是多种细胞周期进程的主要调节因子,它能与相应的调节亚基细胞周期蛋白(cyclin)结合形成有活性的二聚体,参与细胞周期的调节[30]。前期研究表明[31],ALK 抑制剂和CDK 抑制剂联用可以协同降低两条信号通路中p-ALK 和p-Rb 的表达,增强ALK 抑制剂对ALK 的抑制活性及对耐药肿瘤细胞的敏感性,比如在ALK-F1174L 和F1245C 这两种全新耐药突变的异种神经母细胞瘤模型上,即发现CDK4/6 的双重抑制剂瑞博西尼(ribociclib)和ALK 抑制剂色瑞替尼的组合用药会显示出更高的细胞毒性和协同作用。与单用这两种药物相比,联合治疗可增强生长抑制、细胞周期阻滞和促进细胞死亡,并阻止耐药性的出现。
3.1.4 与EGFR 抑制剂联用 EGFR 通路的异常激活是导致肿瘤耐药的重要机制之一。在多种ALK 抑制剂耐药细胞株及患者体内均发现,肿瘤细胞中的NGR1-HER3-EGFR 这条信号通路被代偿性激活,因而肿瘤细胞即使在ALK 信号通路被阻断之后仍可以通过其他信号通路增生分化,继而对ALK 抑制剂产生了耐药[32]。因此,与EGFR抑制剂合用则可以同时阻断这两条信号通路,从而协同发挥抗肿瘤作用,提高治疗效果。例如EGFR 抑制剂阿法替尼(afatinib)与色瑞替尼或者阿来替尼合用,就可以对H3122 色瑞替尼耐药细胞株产生强效抑制作用[36]。此外,在同时具有ALK和EGFR 突变的NSCLC 患者体内也观察到,在使用EGFR 抑制剂厄洛替尼(erlotinib)治疗后,可以使肿瘤细胞对ALK抑制剂也产生部分响应[33]。

3.1.5 与PD-1抑制剂联用 最近几年,免疫治疗已成为抗肿瘤药物的研究热点。研究表明,如果EML4-ALK融合蛋白过表达,会在缺氧或者有氧状态下通过介导缺氧诱导因子HIF-1α和信号传导转录激活蛋白STAT3 的上调,增加程序性死亡受体配体1(programmed death ligand-1,PD-L1)的表达,从而导致T 细胞抑制,致使肿瘤细胞发生免疫逃逸[34]。因而ALK 抑制剂和PD-1/PD-L1 抑制剂联用,已被认为是治疗NSCLC 特别是耐药型NSCLC的一种新选择[35]。目前,多种PD-1/PD-L1 抑制剂如纳武单抗(nivolumab)和Avelumab 与ALK 抑制剂合用来治疗进展性NSCLC 已进入临床研究(NCT02393625,NCT02584634)[36]。
3.2 通过合理设计新型药物分子克服耐药
3.2.1 第4 代ALK 抑制剂 Repotrectinib(TPX-0005),是由美国TP Therapeutics 公司研发的一种具有特殊环状结构的ALK/ROS1/TRK/SRC 小分子抑制剂,也被誉为第4 代ALK 抑制剂[37]。研究表明,Repotrectinib 对多种第1 代、第2 代ALK 抑制剂产生的耐药突变如L1196M 和G1202R 等有效,抑制野生型、L1196M 突变型和G1202R 突变型ALK的亲和力分别为0.87,0.65 和0.81 nmol/L,对野生型ALK 的抑制活性强于克唑替尼,对G1202R 突变型ALK 的抑制活性也强于所有第2 代ALK 抑制剂。Ba/F3 细胞增殖实验也验证了TPX-0005 对各种突变型细胞株具有强效的抑制活性(野生型IC5021.1 nmol/L,L1196M 50 nmol/L 和G1202R 20.5 nmol/L)[38],而且,Repotrectinib 对克唑替尼难治性并且有脑转移的NSCLC 也已被证明了有效[37]。研究表明,Repotrectinib 克服耐药的主要机制是通过抑制SRC/FAK 信号通路,继而抑制肿瘤转录因子YB-1 的磷酸化,导致EGFR、CD44 和波形蛋白的表达下调,最终从多个角度阻碍肿瘤细胞的增生、分化和迁徙[39]。此外,由于其特殊的环状结构可以直接与ATP 结合区结合,而不与溶剂区发生作用,因而可以有效克服G1202R 等一系列突 变[37]。 目 前Repotrectinib 治 疗ROS1、NTRK、ALK 融合实体瘤的Ⅰ/Ⅱ期临床研究正在进行中,登记号NCT03093116,结果令人期待。

3.2.2 Type-Ⅱ型ALK 抑制剂 酪氨酸激酶的晶体结构表明,几乎所有酪氨酸激酶都具有相似的催化结构域。其中,在ATP 结合位点附近,有一段由3 个氨基酸形成的保守活性链段序列,天冬氨酸-苯丙氨酸-甘氨酸(Asp-Phe-Gly,DFG)。DFG 的构象非常关键,当Asp处于ATP结合位点附近的疏水腔外时,激酶处于活性构象(DFG-in),而当其位于激酶的内部时,激酶则处于非活性构象(DFGout)[40]。因此,根据激酶是否处于活性构像,可以将与其结合的抑制剂主要分为:Type-Ⅰ型和Type-Ⅱ型[41]。其中Type-Ⅰ型是指在激酶处于DFG-in构象时,与其ATP 口袋结合的抑制剂,主要属于ATP竞争性的抑制剂。而Type-Ⅱ型则是指在激酶处于非活性构象(DFG-out)时,主要与其变构位点发生结合的抑制剂。此外,还有一种Type-Ⅰ1/2型抑制剂,是指在激酶处于活化构象时,同时也可与Ⅱ型抑制剂主要结合部位发生结合的抑制剂。根据定义,前期开发的第1 代、第2 代抑制剂均属于Type-Ⅰ型或Type-Ⅰ1/2 型抑制剂[42]。 由于Type-Ⅰ型抑制剂主要与ATP 区域的位点结合,而该位点处的氨基酸序列高度保守,导致此类抑制剂容易产生耐药性。而Type-Ⅱ型抑制剂则未完全与ATP 结合位点发生结合,因而虽然其对激酶的亲和力弱于Ⅰ型抑制剂,但是这也提高了它们对激酶的选择性,并且降低了耐药潜力[43],因此开发Type-Ⅱ型抑制剂也成为了获得不易产生耐药的抗肿瘤药物的新选择。Tu等[44]就首次报道了一类新型的以吡唑胺为母核的Type-Ⅱ型ALK 抑制剂,其中化合物5a 对ALK 的体外抑制活性最强(IC50=177 nmol/L)。通过共晶结构分析发现(图2),5a结构中的5-氨基吡唑环仍可以与ALK 的铰链区形成氢键。但与克唑替尼不同的是,其结构中的丁基异唑脲基团会伸展到变构口袋中与变构位点结合,从而诱导ALK 构象发生变化,最终表现出与克唑替尼不同的ALK 结合模式。 该项工作将为Type-Ⅱ型ALK抑制剂的开发提供重要思路。

图2 化合物5a(黄色)、克唑替尼(蓝色)与ALK的共晶复合物叠合图(PDB:2XP2)[44]
3.2.3 其他新型的ALK 抑制剂 为了克服耐药,研究者也设计合成了一些新型的小分子抑制剂。比如针对G1202R 激酶区突变导致的耐药,Mathi等[45]和Geng 等[46]用空间位阻更小的基团如柔性的脂肪胺或者甘氨酸侧链替换了色瑞替尼结构中的哌啶环(化合物10 与12d),增强了化合物与G1202R 突变型ALK 激酶的结合力,增加了疗效,并在一定程度上克服了耐药性。另外,鉴于EGFR与ALK 抑制剂的协同作用,Chen 等[47]也报道了一种ALK/EGFR 双重抑制剂CHMFL-ALK/EGFR-050,该化合物在体内外展现了强效的肿瘤抑制活性。同时,CEP-37440[48],一种ALK/FAK 双靶点抑制剂,也被证实对各种突变型的ALK 激酶均有效,而且该化合物具有更好的脑内药物暴露量,可能对脑转移的肿瘤患者具有潜在的治疗优势,目前该化合物已进入Ⅰ/Ⅱ期临床研究。

3.3 ALK的PROTAC分子
蛋白降解靶向嵌合体(proteolytic targeting chimera,PROTAC)是一类能够通过诱导靶蛋白的多聚泛素化而导致靶蛋白降解的化合物。PROTAC技术作为新兴的药物研发技术,已成为目前肿瘤治疗领域的研发热点。研究表明,通过设计合适的PROTAC 分子可以在一定程度上克服小分子抑制剂的缺陷(例如耐药)[49]。最近,也报道了许多以ALK 为靶蛋白的PROTAC 分子。2018年,Zhang 等[50]以色瑞替尼作为ALK 配体,PEG 作为连接链,来拿度胺作为E3 连接酶配体,报道了ALK 的PROTAC 分子MS4077,结果显示该分子可以以浓度和时间依赖的方式降低SU-DHL-1 淋巴癌和NCI-H2228 肺癌细胞中的ALK 融合蛋白水平。随后,Kang等[51]以VHL 配体替换CRBN 配体,获得了新型的ALK PROTAC 分子TD-004。药理结果表明,TD-004 可以诱导ALK 降解,抑制ALK 融合阳性细胞系SU-DHL-1 和H3122,同时还能有效抑制H3122 异种移植瘤的生长。但需要注意的是,虽然ALK 的PROTAC 分子已有部分报道,并取得了一定进展,但这些分子对耐药细胞的作用还有待进一步检验。
4 总结与展望
ALK抑制剂作为目前ALK阳性NSCLC的主要治疗药物,改善了NSCLC 的治疗模式,延长了广大病患的总生存期和无进展生存期,取得了良好的治疗效果。但是,原发性或获得性耐药的发生,使其在临床上的应用也受到了明显的限制。近年来,随着二次基因突变、旁路激活、上皮间质转化等耐药机制的不断明析,已逐渐开发出针对这些耐药机制的多种治疗策略。例如,设计开发出可以克服基因突变的新1 代ALK 抑制剂或者将ALK抑制剂与多种化疗药物或免疫治疗药物联用。此外,利用PROTAC 技术开发的靶向蛋白降解药物在解决耐药问题方面的潜力也备受关注。然而,虽然这些策略在一定程度上改善了ALK 抑制剂的治疗效果,但均存在着或多或少的不足,比如新型的ALK 抑制剂的研发周期过长、投入大,不能立即满足临床的迫切需求,而联合用药导致的不同药物之间潜在的相互作用以及药代动力学性质差异等问题,不仅会降低疗效,而且可能会增强毒性。另外,PROTAC 药物分子结构过大,水溶性和PK/PD性质不佳,脱靶毒性严重等,也制约着该技术的进一步应用。因此,如何解决现有的关键问题,将很有可能是未来的研究热点和方向。
