18β-甘草次酸的哌嗪酰胺衍生物的合成研究
2020-12-24陈世琴于立权蒙俞帆
陈世琴,于立权,蒙俞帆,汪 婷,蔡 东
(1.锦州医科大学 药学院, 辽宁 锦州 121001;2.营口港务股份有限公司第四分公司,辽宁 营口 115007;3.锦州医科大学公共基础学院,辽宁 锦州 121001)
甘草次酸具有抗肿瘤、抗氧化、抗病毒、抗菌、抗炎、抗过敏、抗动脉粥样硬化的药理活性, 并具保肝解毒活性和类固醇样作作用[1-4],但是临床上的应用常伴有假性醛固酮增多症等副作用,并且甘草次酸几乎不溶于水以及高浓度时对正常细胞的毒性作用,制约了其在临床上的应用。为了减轻、克服这些副作用,科学家们开始致力于改善原料来源丰富的18β-甘草次酸的结构(图1所示)。对18β-甘草次酸的结构修饰主要集中3位羟基、11位羰基和30位羧基结构片段以及A环及C环的结构改造[5]。研究发现[6]18β-甘草次酸的30位羧基酰胺化衍生物的肝靶向性明显强于基本结构甘草次酸。本文通过3位羟基的酯化和30位羧基的酰胺化,对18β-甘草次酸进行修饰,以其得到具有全新结构的甘草次酸衍生物,并对合成过程中副产物产生的条件进行分析,为这类化合物的合理开发和利用提供理论依据。
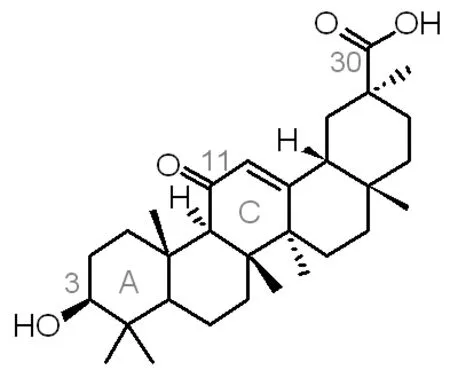
图1 18β-甘草次酸的结构
1 实验步骤
1.1 试剂
18β-甘草次酸(分析纯),2-氯乙酸酐(分析纯)。
1.2 实验步骤
1.2.1 3β-(2-氯乙酰氧基)-甘草次酸(1)的合成
将18 β甘草次酸(0.94 g,2 mmol)、2-氯乙酸酐(3.42 g,20 mmol)和三乙胺(2.55 g,25 mmol)在微波反应器130 ℃回流反应30 min。反应结束后,向反应混合物中趁热慢慢加入50 mL水,搅拌至固体呈粉末状,抽滤、冷水洗得到固体。可用乙醇重结晶得纯品。
化合物(1)为白色粉末状固体,重结晶后收率96.2%; 熔点259.2~261.1℃分解;1HNMR (400 MHz,氘代氯仿) δ 5.72 (s,1H),4.61 (dd,J = 11.8,4.8 Hz,1H),4.06 (d,J = 2.3 Hz,2H),2.83 (m,1H),2.37 (s,1H),2.19 (dd,J = 13.6,4.1 Hz,1H),1.38 (m,3H),1.23 (s,3H),1.17 (s,3H),1.13 (s,3H),0.90 (s,6H),0.84 (s,3H),0.80 (m,1H); HRMS calcd for 547.3190 (C32H48ClO5),found 547.3188。
1.2.2 3β-(2-氯乙酰氧基)-甘草次酸-30-哌嗪酰胺衍生物(3)和(4)的合成
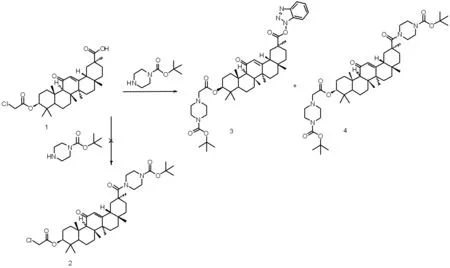
将3β-(2-氯乙酰氧基)-18β-甘草次酸 (0.56 g,1.19 mmol)、1-羟基苯并三唑(HOBT)(0.18 g,1.31 mmol)、 1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC·HCl)(0.25 g,1.31 mmol)和三乙胺(0.14 g,1.31 mmol)加入20 mL乙腈溶液中,室温搅拌30 min。然后再向上述溶液中加入N-叔丁氧羰基哌嗪(0.37 g,2.0 mmol),回流反应10 h,反应结束后减压蒸馏蒸除溶剂,残余物用50%乙醇搅拌均匀后,抽滤,水洗涤后得到固体。硅胶柱层析分离纯化[洗脱剂∶V(乙酸乙酯)∶V(石油醚)=4∶1]后得到18β-甘草次酸的哌嗪衍生物(3)和(4),未见产物(2)。
化合物(3)为白色粉末状固体,纯化后产率10.7%。熔点197.2~199.0 ℃; HRMS calcd for 814.5119 (C47H68N5O7),found 814.5107。
化合物(4)为白色粉末状固体,纯化后产率 75.9%。熔点212.7~213.9 ℃; HRMS calcd for 865.6054 (C50H81N4O8),found 865.6041。
1.2.3 3β-(2-氯乙酰氧基)-甘草次酸-30-酰基哌嗪-1-甲酸叔丁酯(5)的合成
将18β-甘草次酸 (0.56 g,1.19 mmol)、HOBT(0.18 g,1.31 mmol)、 EDC·HCl(0.25 g,1.31 mmol)和三乙胺(0.133 g,1.31 mmol)加入20 mL乙腈溶液中,室温搅拌30 min。然后再向上述溶液中加入N-叔丁氧羰基哌嗪(0.22 g,1.19 mmol),回流反应10 h,反应结束后减压蒸馏蒸除溶剂,残余物用50%乙醇搅拌均匀后,抽滤,水洗涤后得到固体。乙酸乙酯重结晶后得到化合物(5)。化合物(5)为白色固体,重结晶后产率 93.9%。熔点224.3~225.7 ℃;1HNMR (400 MHz,氘代氯仿) δ 5.66 (s,1H),3.63-3.52 (m,4H),3.39 (t,J = 5.2 Hz,4H),3.22-3.18 (m,1H),2.79-2.74 (m,1H),2.31 (s,1H),2.30-2.23 (m,1H),1.45 (s,9H),1.34 (s,3H),1.20 (s,3H),1.11 (s,3H),1.10 (s,3H),0.98 (s,3H),0.79 (s,3H),0.78 (s,3H),0.68 (d,J = 11.6 Hz,1H); HRMS calcd for 639.4737 (C39H63N2O5),found 639.4736。
1.2.4 3β-(2-氯乙酰氧基)-甘草次酸-30-哌嗪酰胺衍生物(6)和(7)的合成
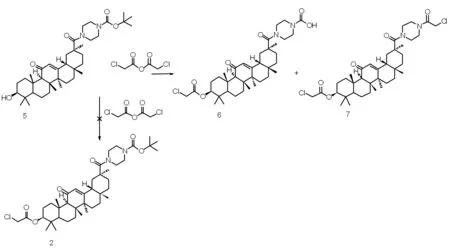
化合物(5)(1.28 g,2 mmol)、2-氯乙酸酐(3.42 g,20 mmol)和三乙胺(2.55 g,25 mmol)在微波反应器130 ℃回流反应30 min。反应结束后,向反应混合物中趁热慢慢加入50 mL水,搅拌至固体呈粉末状,抽滤、冷水洗得到固体。硅胶柱层析分离纯化[洗脱剂∶V(二氯甲烷)∶V(甲醇)=50∶1~10∶1,梯度洗脱]后得到18β-甘草次酸的Boc-哌嗪衍生物(6)和(7),未见产物2。
化合物(6)为白色固体,产率 22.4%。熔点205.7.0~206.6 ℃;1HNMR (400 MHz,氘代氯仿) δ 5.66 (s,1H),4.59 (dd,J = 11.8,4.7 Hz,1H),4.15 - 3.97 (m,4H),3.65 (s,4H),3.52 (s,2H),2.85 - 2.75 (m,1H),2.34 (s,1H),2.28 (d,J = 12.1 Hz,1H),1.34 (m,3H),1.22 (s,3H),1.14 (s,3H),1.10 (s,3H),0.88 (s,6H),0.80 (s,3H),0.78 (m,1H); HRMS calcd for 659.3827 (C37H56ClN2O6),found 659.3873。
化合物(7)为白色固体,纯化后产率 76.7%。熔点177.4~175.7 ℃;1HNMR (400 MHz,氘代氯仿) δ 5.67 (s,1H),4.60 (dd,J = 11.8,4.7 Hz,1H),4.14-3.99 (m,4H),3.73-3.66 (m,4H),3.66 - 3.55 (m,2H),3.53 (d,J = 5.4 Hz,2H),2.81 (dt,J = 13.8,3.7 Hz,1H),2.35 (s,1H),2.30 (dd,J = 13.4,3.8 Hz,1H),1.35 (m,3H),1.23 (s,3H),1.15 (s,3H),1.11 (s,3H),0.89 (s,6H),0.81 (s,3H),0.79 (m,1H); HRMS calcd for 691.3645 (C38H57Cl2N2O5),found 691.3641。
2 实验结果与讨论
我们最初设想以18β-甘草次酸为起始原料,经3位羟基发生酯化反应,然后化合物(1)的30位羧基和N-叔丁氧羰基哌嗪发生酰胺化反应得到化合物(2)。为此,我们投入过量的N-叔丁氧羰基哌嗪与化合物(1)反应,并利用HOBT、EDC·HCl和三乙胺作为活化剂,反应物纯化后主要得到化合物(3)和(4),未见产物(2)。而且化合物(3)和(4)的产率比例随着N-叔丁氧羰基哌嗪投入量的增加而改变。这可能是由于化合物(1)靠近30位羰基的取代基空间位阻很大,使得N-叔丁氧羰基哌嗪无法接近继而发生亲核加成反应。另外,化合物(1)结构中的3β-2-氯乙酰氧基结构片段在乙腈溶液中(三乙胺存在下)极易和N-叔丁氧羰基哌嗪中N原子发生亲核取代反应。也就是说该反应条件下,化合物(1)的30位羧基片段和3β-2-氯乙酰氧基结构片段均可发生反应,很难得到设想的单一产物(2)。
为此,我们改变反应顺序,希望得到产物(2)。参照化合物(3)和(4)的合成条件,首先合成化合物(5),再和2-氯乙酸酐反应。但是反应过程中形成的弱酸2-氯乙酸,不能立即、完全和三乙胺发生中和反应,反应体系中残存的2-氯乙酸可以选择性地水解哌嗪-1-甲酸叔丁酯结构片段。随着反应温度和反应时间的不同,产物比例也有差异。可能的反应机理如下图2所示。
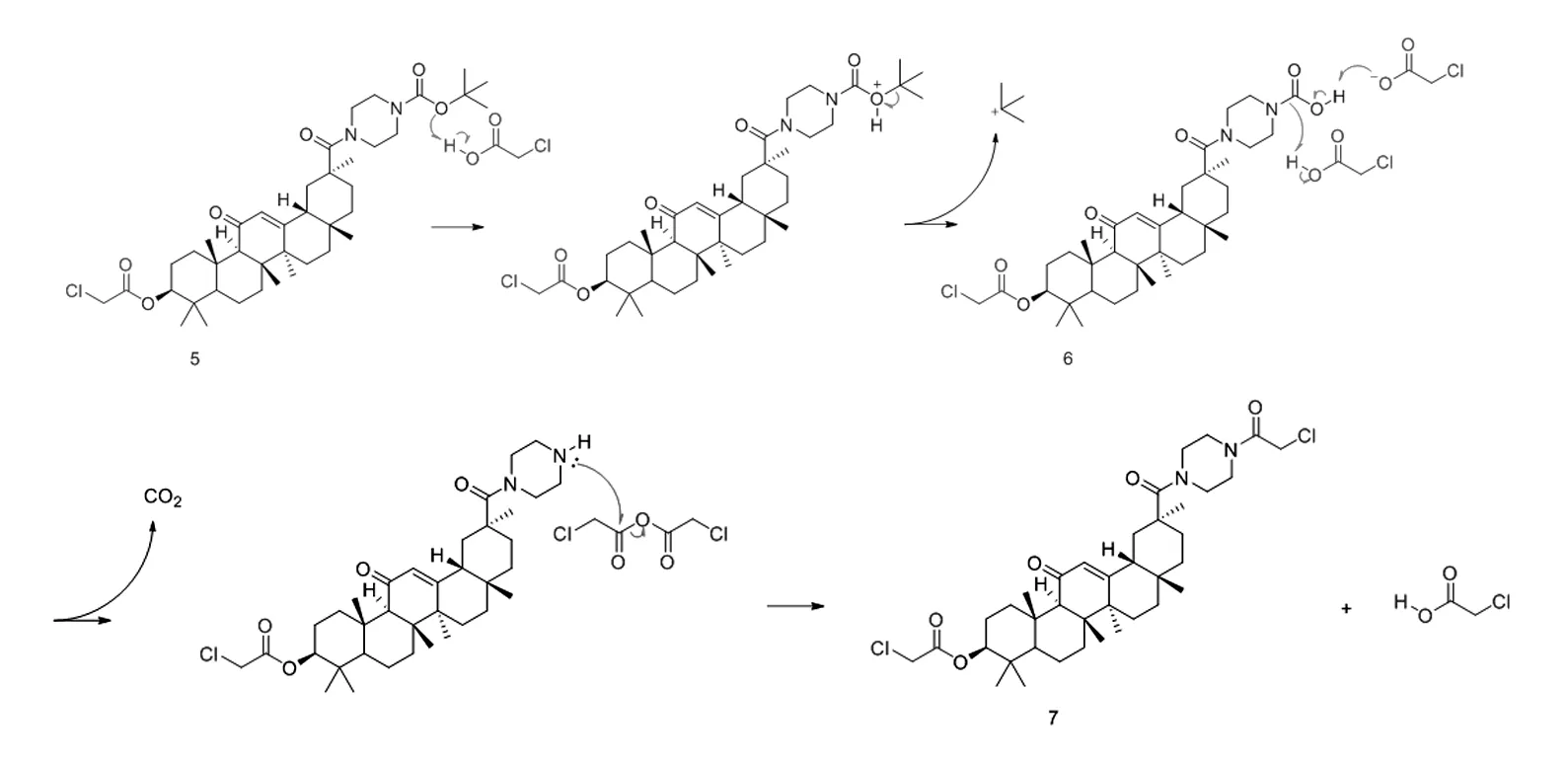
图2 化合物(2)的水解过程
3 结论
本实验未能制备3β-(2-氯乙酰氧基)-18β-甘草次酸-30-酰基哌嗪-1'-甲酸叔丁酯(2)。但合成了化合物(3)、(4)、(6)、(7),并分析了生成的原因,为18β-甘草次酸的合理开发提供了新的候选化合物。
