基于UPLC-MS/MS测定烘焙咖啡豆中美拉德产物F3-A含量
2020-12-23王东旭胡奇杰王凤丽王新财厉芬陈褚建
王东旭,胡奇杰,王凤丽,王新财,厉芬,陈褚建
(湖州市食品药品检验研究院,浙江 湖州,313000)
美拉德反应(Maillard reaction)是一种在食品热加工和贮藏过程中广泛存在的非酶褐变反应,主要是指食品中的羰基化合物(还原糖类)和氨基化合物(氨基酸和蛋白质)间发生的复杂反应,又称羰胺反应[1]。随着美拉德反应的进行,会产生一系列复杂的化合物,称为美拉德反应产物(Maillard reaction products,MRPs)。MRPs广泛存在于各种食品体系中,对食品的色泽、风味、稳定性和营养价值具有很大的影响[2-3]。[5-(5,6-dihydro-4H-pyridin-3-ylidenemethyl)-furan-2-yl]methanol,简称F3-A,是葡萄糖-赖氨酸反应模型中发现的一个美拉德反应产物[4-7]。药理实验研究显示,F3-A在细胞因子诱导的Caco-2细胞中对NO及诱导性NO合成酶具有明显的抑制作用,是一种潜在的预防肠道炎症活性物质[8]。Caco-2细胞渗透性实验表明,F3-A还具备较好的渗透性,同时由于F3-A具备良好的水溶性,其具有较高的生物利用度,因此有望在体内发挥抗炎作用[9]。
F3-A作为美拉德反应模型产物,主要由葡萄糖脱水形成的5-羟甲基糠醛与赖氨酸经Strecker降解、脱水环化形成的2,3,4,5-四氢吡啶发生缩合反应形成[9]。但其在真实食品中的研究还较少,目前文献报道只在烘焙面包中检出[9],在其他食品中的研究还未见报道。
咖啡中含有大量的活性成分,具有减肥、提神醒脑、利尿、抗氧化等保健功效[10-12],其中绿原酸[13-14]、咖啡因[15-17]、葫芦巴碱[16-19]等已得到较多研究,但烘焙咖啡豆中F3-A含量的检测尚未见报道。基于对F3-A的生成机理分析,咖啡生豆中含有约60%糖类物质和0.6%赖基酸[20],在烘焙环境下,推测烘焙咖啡豆中会有F3-A生成。同时目前关于F3-A的检测方法主要为液相色谱法[9],该法具有对复杂基质分离要求较高,检出限相对较高等不足。超高效液质联用法(ultra-performance liquid chromatography-tandem mass spectrometry,UPLC-MS/MS)作为一种集液相色谱的高分离效能与质谱的强鉴定能力于一体的检测技术,具有适用范围广、灵敏度高、重复性好的特点,因而开发针对烘焙咖啡豆中F3-A的液质联用方法对于探究F3-A在咖啡豆中的存在与否具有重要意义,此项研究同时有助于开发咖啡的潜在药用价值和经济价值。
本研究以5种咖啡豆(分别产自洪德拉斯、肯尼亚、印尼、巴西和萨尔瓦多)为原料,经过不同程度(轻度、中轻度、中度、中深度、重度和重深度)的烘焙,采用UPLC-MS/MS法检测F3-A含量。通过分析F3-A含量变化,揭示不同烘焙时间、温度对不同品种咖啡豆F3-A成分的影响,为调控烘焙咖啡豆中F3-A含量、提高咖啡药用价值提供一定的理论依据。
1 材料与方法
1.1 材料与仪器
标准品: [5-(5,6-dihydro-4H-pyridin-3-ylidenemethyl)-furan-2-yl]methanol(F3-A),按参考文献 [6]合成,结构经红外谱图和质谱确证,经高效液相色谱峰面积归一化法定量测定,纯度>95%;甲醇(色谱纯),德国Merck公司;甲酸(色谱纯)、氨水(分析纯,质量分数25%~28%),上海麦克林生化科技有限公司;盐酸(分析纯,质量分数36.0%~38.0%),江苏强盛功能化学股份有限公司;Waters Oasis MCX(混合型强阳离子交换反向吸附剂)固相萃取小柱(60 mg,3 mL)、Waters Oasis WCX(混合型弱阳离子交换反相吸附剂)固相萃取小柱(60 mg,3 mL)、Waters Oasis HLB(全能型亲水亲脂平衡反相吸附剂)固相萃取小柱(60 mg,3 mL),美国Waters公司;试验用水为超纯水。
咖啡原料:洪德拉斯SHB咖啡豆,产自洪都拉斯NINFA LANZA庄园;肯尼亚AA++咖啡豆,产自肯尼亚奇雅瓦姆鲁鲁;印尼罗布斯塔咖啡豆,产自印度尼西亚爪哇;巴西喜拉多咖啡豆,产自巴西喜拉多;萨尔瓦多红波旁咖啡豆,产自萨尔瓦多Loma La GIoria庄园。
ACQUITYTM 超高效液相色谱、XevoTMTQD三重四极杆串联质谱仪,美国Waters公司;ME204E型电子分析天平,瑞士Mettler Toledo公司;Genius 3漩涡混合器,德国IKA公司;KB-5010型试管振荡器,海门市其林贝尔仪器制造有限公司;KQ-500DB型超声清洗仪,昆山市超声仪器有限公司;Allegra X-12R型台式冷冻离心机,美国Beckman Coulter公司;12孔固相萃取仪,美国Supelco公司;Turbovap LV型多样品自动浓缩仪,瑞典Biotage公司;Simplicity型超纯水仪,美国MilliPore公司;L9/11/B410型马弗炉,德国Nabertherm公司;JFSD-100型粉碎机,上海嘉定粮油仪器有限公司。
1.2 溶液的制备
1.2.1 对照品溶液的制备
准确称取F3-A标准物质适量,用甲醇溶液配制成质量浓度为1.00 mg/mL储备溶液,置于-20 ℃冰箱中保存。根据需要移取标准储备溶液,经初始流动相溶液配制成质量浓度分别为5、10、50、100、500和1 000 ng/mL的标准工作溶液。取1.00 mL标准溶液经0.22 μm有机相滤膜过滤后供UPLC-MS/MS检测。
1.2.2 供试品溶液的制备
随机选取咖啡豆平铺一层于坩埚盘中,放入马弗炉固定位置烘焙。设定烘焙温度为220 ℃,烘焙时间分别为10、20、30、40、50和60 min,烘焙完成后,立即取出冷却至室温。参照文献[18]按咖啡豆爆裂程度定义为轻度(一爆密集爆)、中轻度(一爆尾爆)、中度(一爆结束二爆前)、中深度(二爆初爆)、重度(二爆密集爆)、重深度(二爆尾爆)。收集烘焙咖啡豆样品,用粉碎机将咖啡豆粉碎(粒径1.00 mm),供含量测定。
称取1.00 g(精确至0. 01 g)咖啡豆粉置于15 mL离心管中,加入10.00 mL 1 mol/L HCl溶液,涡旋2 min,超声20 min,以9 000 r/min离心5 min,取上清液5.00 mL过固相萃取小柱(依次用3.00 mL甲醇和3.00 mL水活化),经3.00 mL水和3.00 mL甲醇淋洗,6.00 mL 体积分数5%氨水甲醇溶液洗脱,收集洗脱液于试管中,40 ℃下氮吹至近干,加入1.00 mLV(甲醇)∶V(0.1%甲酸水溶液)=5∶95混合溶液复溶,混匀,过0.22 μm有机滤膜。按仪器工作条件进行测定。
1.3 测定方法
分别精密吸取上述不同浓度对照品溶液各5.00 μL,注入高效液相串联质谱仪,测定出峰面积,以质量浓度(ng/mL)(X)为横坐标,峰面积(Y)为纵坐标,绘制F3-A化合物标准曲线图。精密吸取上述供试品溶液5.00 μL,注入高效液相串联质谱仪,测定峰面积,采用外标法定量。
1.4 色谱及质谱条件
色谱柱Waters ACQUITY UPLC BEH C18(100 mm×2.1 mm,1.7 μm);流动相A为甲醇溶液,流动相B为体积分数0.1%甲酸水溶液,梯度洗脱程序:0.0~1.0 min,5%A;1.0~2.0 min,5%~50%A;2.0~3.0 min,50%~95%A;3.0~4.0 min,95%A;4.0~4.1 min,95%A~5%A;4.1~7.0 min,5%A;流速0.30 mL/min;进样体积5.00 μL;柱温40 ℃。
质谱条件:电喷雾离子源(electron spray ionization,ESI)电离,正离子多反应监测(multiple reaction monitoring,MRM)模式检测;毛细管电压为5.0 kV;离子源和脱溶剂气温度分别为150和350 ℃;脱溶剂气和锥孔气流速分别为800和50 L/h;碰撞气为氩气;F3-A的定性定量离子对(m/z)、锥孔电压及碰撞能量等质谱参数见表1。

表1 F3-A的监测离子对及最佳质谱参数Table 1 Selected transitions and optimized potentials of F3-A
2 结果与分析
2.1 前处理条件的优化
由于咖啡豆基质较为复杂,采用振荡提取和液液萃取作为提取和纯化方法提取效率不高,纯化效果不佳[9]。因此本研究分别对提取和纯化方式进行了优化。在其余条件完全一致情况下,分别采用振荡提取20 min和超声20 min,考察2种方式的提取效率。结果表明,超声提取得到的目标物响应面积较振荡高(15.2±1.3)%,故确定超声20 min作为最佳提取方法。其次考察了各种固相萃取小柱的纯化效果,由于F3-A为弱碱性,本研究选取了Oasis HLB、Oasis MCX、Oasis WCX 3种相同品牌及规格,但不同填料类型的固相萃取小柱进行纯化优化,如图1所示。结果显示,WCX和MCX均能有效地对F3-A进行富集及纯化,其中MCX的富集及纯化效果最佳,而HLB则较难在柱中保留,故最终选取MCX作为最优固相萃取小柱。
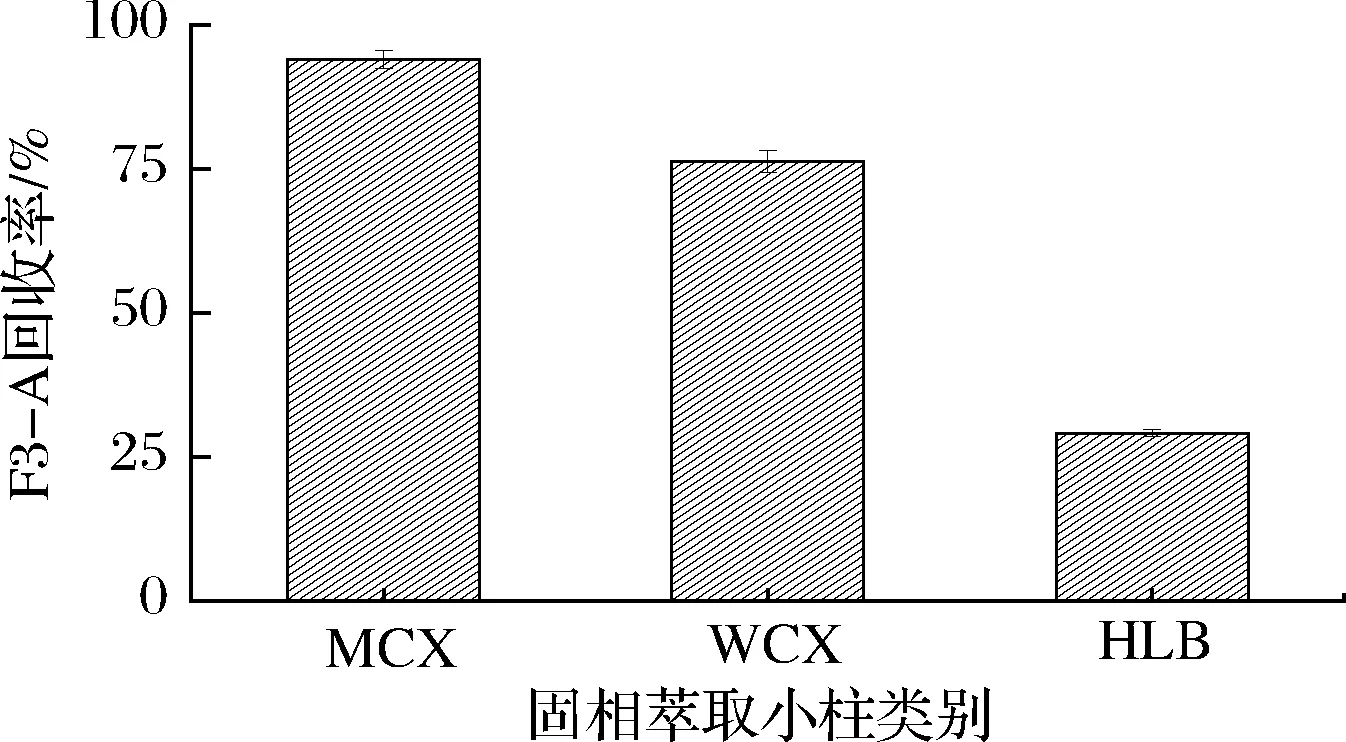
图1 固相萃取小柱优化Fig.1 Optimization of solid phase extraction column
2.2 标准曲线、线性范围和检出限
将F3-A对照溶液在上述液相质谱条件下进行测定,绘制进样浓度为横坐标、定量离子对峰面积为纵坐标的F3-A化合物标准曲线,线性拟合得到线性方程和决定系数,结果见表2。F3-A线性拟合良好,决定系数达到0.999。F3-A对照品溶液色谱图及阳性样品色谱图见图2。F3-A峰形良好,目标峰周围无杂峰干扰,满足定量检测要求。根据信噪比(S/N)=3,采用不含F3-A的空白基质样品提取液逐级稀释F3-A标准溶液分析得到检出限。
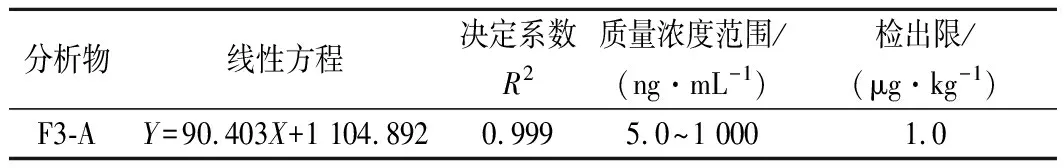
表2 回归方程和线性范围Table 2 Linearity Parameter and linear range of F3-A
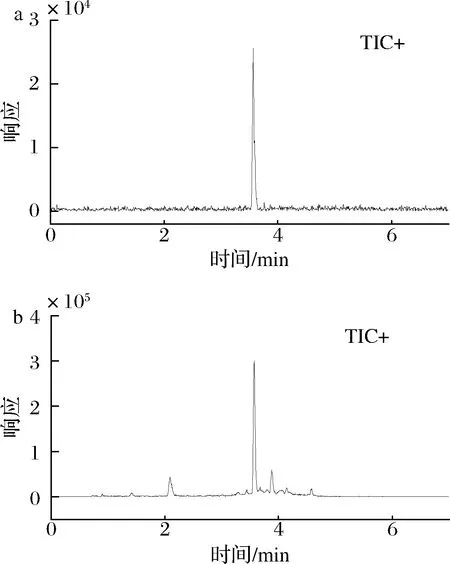
a-F3-A标准溶液色谱图(10 ng/mL);b-阳性样品色谱图图2 F3-A标准溶液色谱图和阳性样品色谱图Fig.2 Chromatograms for the standard solution of F3-A and the positive sample
2.3 回收率和精密度实验
在咖啡豆样品中分别加入3个浓度梯度的F3-A标准溶液,按上述条件平行测定6次,扣除本底含量后计算加标回收率和相对标准偏差,结果见表3。采用该方法测定F3-A的加标回收率为85.7%~93.3%,相对标准偏差为2.56%~3.97%。F3-A化合物精密度和回收率均满足定量检验需求。

表3 F3-A精密度与回收试验结果(n=6)Table 3 Test results for recovery and precision(n=6)
2.4 烘焙咖啡豆F3-A含量检测及调控
基于新建立的咖啡豆中F3-A的液质联用检测方法,在220 ℃下对5种咖啡生豆中F3-A含量进行了测定,结果见图3。未烘焙情况下,5种咖啡生豆中均未检出F3-A,随着烘焙条件的加入,正如之前推测,5种咖啡豆中均检测出F3-A,随着烘焙时间的增加,F3-A的含量先升高再下降,烘焙10 min时,除了萨尔瓦多红波旁咖啡豆,其余4种咖啡豆中F3-A含量达到峰值,肯尼亚AA++咖啡豆含量最高达到297.5 μg/kg。烘焙时间继续增加,F3-A含量则逐渐下降,烘焙时间达到40 min时,5种烘焙咖啡豆中F3-A含量不再检出。因此,确定10 min为最佳咖啡豆烘焙时间。
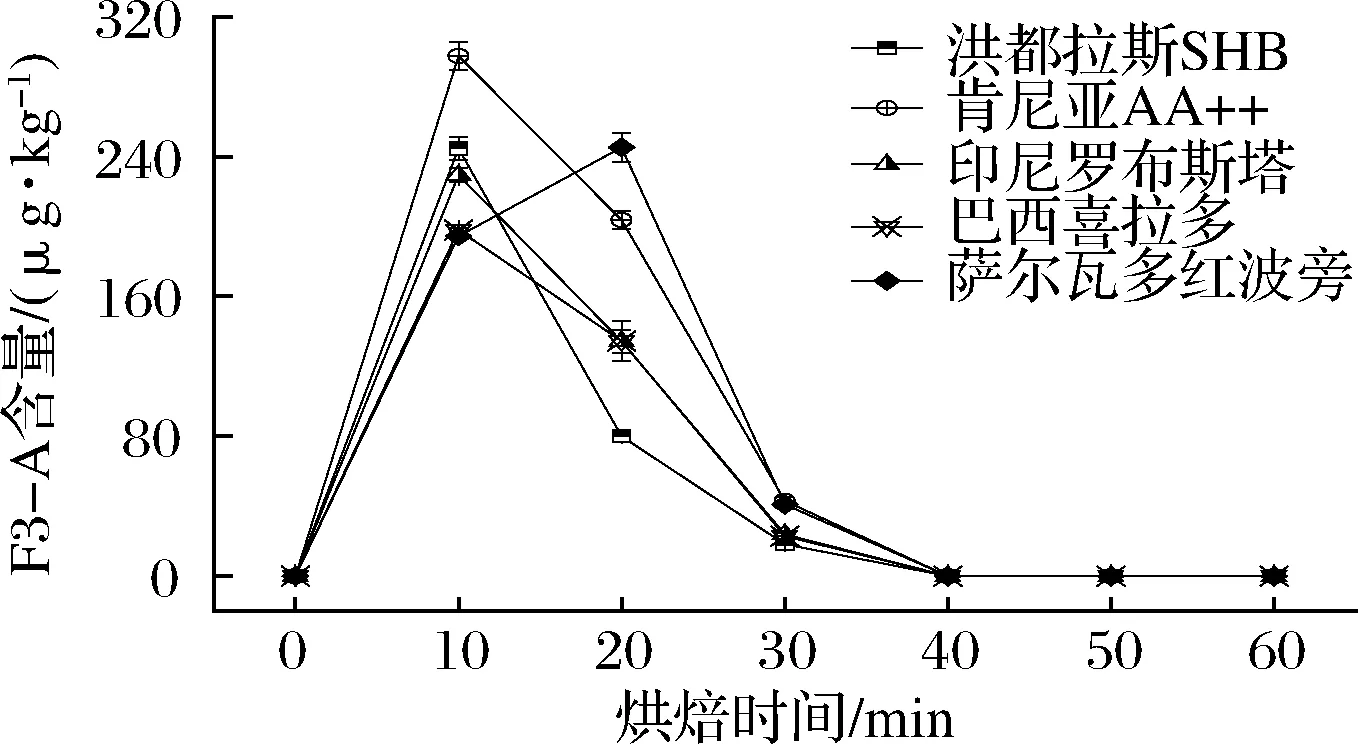
图3 五种咖啡豆不同烘焙时间下的F3-A含量变化(n=3)Fig.3 F3-A content of 5 coffee beans with different roasting time (n=3)
不同烘焙温度下烘焙10 min后对5种咖啡豆中F3-A的含量进行测定,结果见图4。不同烘焙温度下F3-A含量差异较大, 200 ℃时,5种咖啡豆中F3-A含量达到峰值,洪都拉斯SHB和肯尼亚AA++咖啡豆含量最高,分别达到了318.1和329.5 μg/kg,温度继续增高会造成F3-A含量下降。因此最终确定咖啡豆最佳烘焙温度为200 ℃。
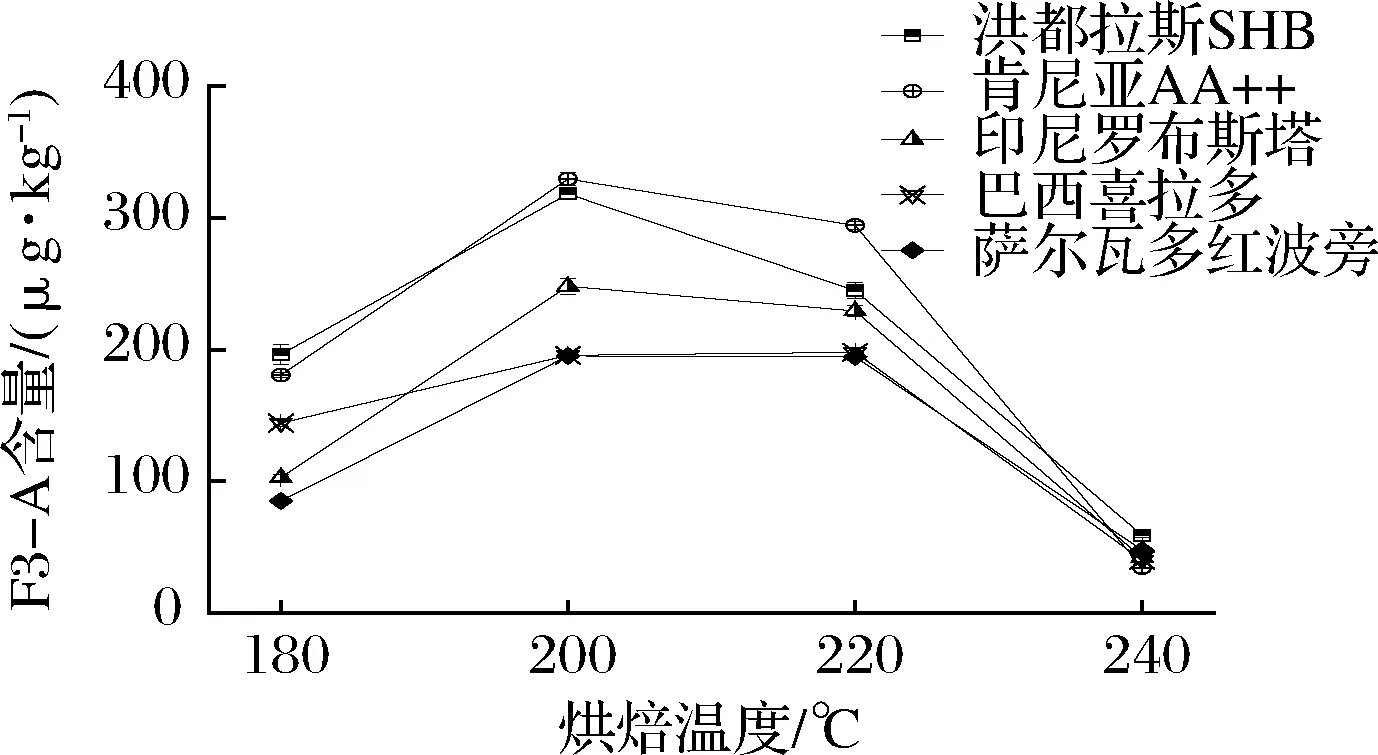
图4 五种咖啡豆不同烘焙温度下的F3-A含量变化(n=3)Fig.4 F3-A content of 5 coffee beans with different roasting temperature (n=3)
结合不同温度和不同时间下F3-A含量比对发现,二者之间存在交叉影响,咖啡豆在240 ℃烘焙10 min与220 ℃下烘焙30 min两种模式下,F3-A含量接近,表明低温长时烘焙和高温短时烘焙具有类似的烘焙效果,其原因推测为F3-A分解速率受温度因素影响所致,在较高温度下F3-A分解越快,较低温度下F3-A含量维持更久。
3 结论
本文建立了以UPLC-MS/MS检测咖啡豆中F3-A含量的检测方法,采用盐酸溶液提取、MCX固相萃取小柱纯化,在MRM模式下检测,外标法定量。该分析方法灵敏度好、准确度高、实用性强,其中检出限为1.0 μg/kg,远低于已报道[9]的21.0 μg/kg,为咖啡豆中F3-A含量检测尤其是痕量检测提供了技术支持。本文首次发现了烘焙咖啡豆存在美拉德产物F3-A,且F3-A含量与烘焙条件和咖啡豆品种密切相关。不同烘焙温度及时间下,5种咖啡豆中F3-A含量测定结果显示,轻度烘焙时F3-A含量普遍较高,最优烘焙条件为200 ℃下烘焙10 min。肯尼亚AA++咖啡豆较其他4种咖啡豆中生成F3-A含量更高,达到329.5 μg/kg。与文献[9]报道烘焙面包中F3-A的最高含量(177 μg/kg)相比,烘焙咖啡豆中F3-A含量更高,更具有潜在的药用价值。
