BaBiO3电子结构与机械性能的第一性原理研究
2020-12-18王殿辉胡朝浩
刘 林, 王殿辉, 钟 燕, 胡朝浩
(桂林电子科技大学 材料科学与工程学院,广西 桂林 541004)
目前,作为一种新型高温超导材料,BaBiO3吸引了科研人员的广泛关注。BaBiO3存在单斜、菱方和面心立方结构,并可以发生相转变。在常温状态下,BaBiO3以单斜I2/m相存在,当温度达到425 K时,单斜I2/m相转变成菱方R-3相,直到温度达到893 K时再次发生相变,转变为面心立方结构[1]。早期,科研工作者在BaBiO3中加入钾元素,发现可以形成具有高于30 K超导温度的化合物Ba1-xKxBiO3[2]。由于该化合物中Bi具有+3和+5的混合价态而产生的电荷歧化,极有可能是获得高温超导的原因[3]。BaBiO3具有三维的Bi-O阵列结构,与传统的铜基超导体二维片层结构完全不同。Tang等[4]的研究表明,BaBiO3中Bi原子存在+3、+5价,且这2种价态的Bi原子对光催化性能的贡献不同。鉴于此,为了探明BaBiO3的超导机理和光催化性能,开展研究Bi混合价态的工作是必不可少的。为此,运用第一性原理计算了BaBiO3三种相的电子结构,并在此基础之上通过用钽(Ta)掺杂BaBiO3改变电子能带结构和BaBiO3机械性能,为今后该材料的实验提供理论依据。
1 计算方法
BaBiO3与Ta掺杂BaBiO3的电子结构和力学性能均在VASP软件包[5]中完成。原子核与电子的交互作用采用投影缀加波法(PAW)[6-7]来描述,交换关联能采用perdew-burke-ernzerhof(PBE)修正的广义梯度近似(GGA)来处理[8]。为了得到精确的晶体结构,对初始结构进行充分的驰豫,其截断能设置为520 eV,采用不大于2π×0.03 Å-1的k点网格密度。在优化过程中,当能量的收敛度小于1×10-5eV,且每个原子受力小于0.01 eV/Å时,结构优化完成。为了快速准确地得到计算结果,在整体计算过程中,用费米形式的展宽模式来计算电子态密度。
2 结果与讨论
2.1 BaBiO3的晶体结构
表1为BaBiO3三种相的优化晶格常数,图1为经过结构优化后BaBiO3的立方、菱方、单斜相晶体结构。3种晶相的BaBiO3由BiO6八面体结构组成,其中Bi原子位于八面体中心,O原子位于八面体各顶角。在BiO6八面体结构单元中,由于Bi原子以+3和+5混合价态存在,导致出现2种Bi—O键键长。八面体与八面体之间通过共享一个O原子相连接,并形成电子传输通道。

表1 BaBiO3三种相的优化晶格常数

图1 BaBiO3的晶体结构
在立方和菱方的结构中存在2种不同的Bi—O键长,在—Bi—O—Bi—O—Bi—键所形成的平面上,O原子的排列具有疏密性偏离Bi原子的现象,这个现象从电荷密度图中也可以观察到[10]。BaBiO3三种结构优化前后Bi—O键长的变化列于表2中,其中Bi(I)表示+5价Bi原子,Bi(II)表示+3价Bi原子。从表2可看出,BaBiO3面心立方结构和菱方结构在结构优化前后,2种Bi—O键长差均发生改变,面心立方中Bi—O键的键长差减小到0.041 3 Å,但在菱方结构中反而增大到0.158 0 Å。对于单斜结构来说,优化前后Bi—O键的键长变化较为复杂,八面体中的Bi—O键长不相等。从表2可看出,存在2种不同长度的Bi—O键,较长的Bi—O键长为2.32 Å,而较短的Bi—O键长约为2.17 Å。一个八面体中的Bi—O长键通过顶角的O原子与紧邻的八面体中的Bi—O短键互连。由于键长的方向依赖性产生对应的电荷方向依赖性,导致了单斜结构的产生。值得注意的是,光生电子空穴的传输,光催化性能和材料的各向异性均受这种特性的影响。
在单斜结构对称的体系中,存在一种Bi—O“breathing distortion”(呼吸形变)模式[11],这种模式不是普遍存在的,而是由单斜结构八面体中Bi原子电荷的歧化造成的。在表2中的实验数据[9]展示了这2类八面体的呼吸形变为同一八面体中不同类型Bi-O的差值,即为0.000 3 Å和0.008 5 Å,与本次计算值0.003 7 Å和0.009 1 Å基本相符。由此可表明,这种呼吸模式影响催化剂的催化性能、自身相结构或者晶面的性质。如BiVO4单斜结构的光催化性能在3种结构中最高;另外,在BiVO4和金红石TiO2的各个晶面中,(110)面的氧化能力最强。

表2 具有不同对称性结构的BaBiO3键长分布 Å
2.2 BaBiO3的电子结构
在得到优化结果的基础上,进一步计算了BaBiO3的能带结构和电子态密度。对于DFT计算,往往只关注靠近费米能级附近的几条能带。BaBiO3三种晶体结构的能带结构如图2所示。

图2 BaBiO3三种不同晶体结构的能带图
相关BaBiO3立方结构的实验结果表明,其为间接半导体,但测得的带隙值差别较大,变化范围在0.2~2.0 eV[12-16]。由于在正常的DFT计算中未考虑电子的激发效应,计算结果一般比实验值偏小。从图2(a)可看出,因面心立方结构的BaBiO3的带隙被低估,导致了价带与导带重叠。BaBiO3菱方和单斜结构分别在高对称点L、V处为最小带隙,被低估后的带隙分别为0.20、0.05 eV,且与BaBiO3的立方结构一样,均为间接半导体。
为了更深入地研究BaBiO3的电子结构,以BaBiO3立方结构为例进行电子态密度分析。Bi(I)为+5价,Bi(II)为+3价的立方结构BaBiO3态密度图如图3所示。从图 3可看出,BaBiO3的电子态密度贡献主要分布在3个区间,在-1~0 eV区域的态密度主要由Bi(II)—6s、O—2p及少量的Bi(I)—6p组成。在0~2 eV附近电子态密度的主要有Bi(I)—6s、Bi(II)—6p、O—2p及少量的Bi(I)—6p、Bi(II)—6s组成。在能量大于2.5 eV的区间电子态密度主要由Ba—5d、Bi(I)—6p、Bi(II)—6p、O—2s及O—2p组成。由于Bi(I)和Bi(II)在离子状态下电荷量不同,2种Bi原子在费米能级附近的区间分布也不同。Bi(I)因为与O—2p形成σ反键态,所以产生了Bi5+离子。此时Bi(I)—6s轨道无电子占据,为空轨道,所以Bi5+离子分布于费米能级之上的占据态;同样,Bi(II)中的6s与O—2p形成σ成键态,因此产生了Bi3+离子,分布区间在费米能级以下。从图2(a)可看到一条贯穿费米能级的能带,这是由费米能级附近Bi—6s和O—2p杂化效应造成的。

图3 立方结构BaBiO3态密度图
2.3 Ta掺杂BaBiO3的电子结构
具有典型ABO3型钙钛矿结构的BaBiO3由2种不同价态的Bi原子周期性排列,形成明显的层状结构。对于50% 的Ta掺杂,由于周期性中心和棱占位,中心和面心的位置是完全等效的。也就是说,沿a、b、c轴中任意方向移动晶格长度的一半,得到的掺杂模式能带结构与移动前是一致的,能带图如图4(a)所示,带隙约为2.66 eV。 若不考虑BaBiO3的对称性,则只在同一个层状结构中替换Bi而掺入50%的Ta原子。这样仅保证了该掺杂结构具有50% Ta的掺杂量。该结构破坏了原有的周期性,导致该掺杂模型的能带结构中价带与导带相交,表现出金属性,如图4(b)所示。因此,掺杂Ta的位置对材料本身的性能影响较大。
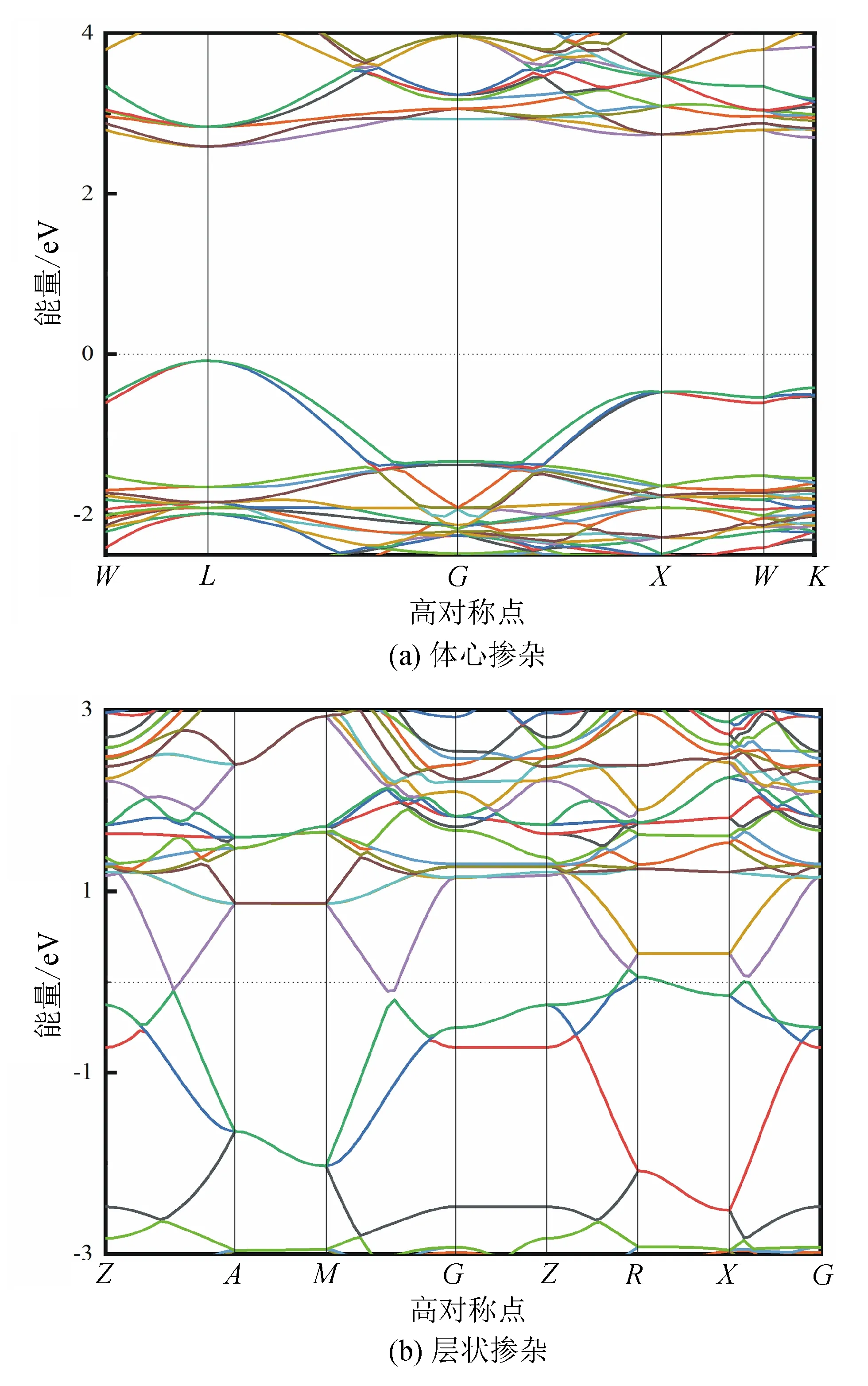
图4 Ta替换Bi掺杂浓度为50%的BaBiO3能带结构
当Ta替换Bi的掺杂浓度为12.5%时,Ta的掺杂在面心立方的体心、面心或者棱中点时的能带结构是一致的,如图5(a)所示。因为晶格的对称性,该掺杂浓度下,不管是体心掺杂模型,还是面心掺杂模型、棱掺杂模型,都可以通过沿着晶格轴平移来相互转换。例如,体心掺杂移动a/2的长度就会转换为面心掺杂模型,然而整个晶格的对称性却未发生变化。通过观察能带结构可发现,价带与导带相交,表现为半金属特征。与纯相BaBiO3的能带结构相比,12.5% Ta掺杂的能带结构中,使低能带的离域性和高能空带的定域性都获得了加强。由此可推断出,从纯相BaBiO3开始不断提高Ta的掺杂浓度,直到浓度达到12.5%的这个过程中,费米能级附近能级较低的能带会向高能级方向移动。掺杂浓度为37.5% Ta的情况与此浓度情况类似,所有掺杂模型的能带结构完全相同,如图5(b)所示。

图5 Ta替换Bi掺杂浓度分别为12.5%、37.5%的BaBiO3能带结构
通过掺杂方式的不同,研究了2种不同的25% Ta掺杂模型。一种用Ta原子在面心立方中的面上替换2个Bi原子,掺杂后的结构如图6(a)所示;另一种掺杂方式是使用Ta替换面心立方中一个面心Bi原子和一个体心Bi原子,结构如图6(b)所示。从图6可看出,2种掺杂结构均表现出金属性,掺杂结构中的Bi具有混合价态,仍能出现Bi的电荷歧化现象。从能带结构还可分析出,采用合适的能级修正,在图6(a)中的高对称点A和图6(b)中的高对称点G均可能出现直接带隙。

图6 Ta替换Bi掺杂浓度为25%的BaBiO3能带结构
对使用Ta替换Bi不同浓度的掺杂后,能带结构计算结果表明,仅有不破坏对称性的50% Ta会显示出明显带隙,其他12.5% Ta、25% Ta和37.5% Ta的3种浓度掺杂后均无带隙,表现出金属性。另外,从能带结构可看出,能带越过费米能级后,未发生严重的交叉。这2种现象可能是由混合价态(+3,+5)的Bi原子电荷歧化导致的。为了进一步探究其本质原因,利用杂化泛函对能带进行了修正,但依然由于Bi原子电荷的歧化现象,处理仍比较困难。
2.4 BaBiO3的力学性质
BaBiO3具有立方、三方(菱角形式)、单斜3种不同的晶系,其对应的独立弹性常数的个数也不尽相同。其中立方晶系具有3个独立的弹性常数,计算结果如表3所示。对于立方晶系,相应的力学稳定性条件为:C11>0,C44>0,C11>C12,(C11+C12)>0。通过稳定性条件的验证,立方晶系BaBiO3满足上述判定条件。

表3 计算得到的BaBiO3的弹性常数Cij、体模量B、剪切模量G、杨氏模量E及泊松比υ
材料抵抗正应力的能力和抵抗剪应力的能力分别用体模量B和剪切模量G来表示。其中B/G与材料的脆性相关,若B/G>1.75,则表明该材料具有易延展性[17]。立方结构BaBiO3的B/G=1.98,表现出良好的延展性。若材料本身的各向异性比较微弱,则可以用E=9BG/(3B+G),υ=(3B-2G)/2(3B+G)来近似求解各向同性材料的杨氏模量E与泊松比υ[18]。泊松比υ用来表示材料在形变过程中体积的改变程度,其值越小,该材料在形变过程中体积改变越大。立方结构的BaBiO3的泊松比为0.28,表明具有较好的韧性。根据泊松比υ,还可以从力学角度得到更多的成键方面的信息[19-20]。另外,根据Zener定义[21],可用参数A来描述立方结构BaBiO3的各向异性,其中A=G1/G2,G1=C44,G2=(C11-C12)/2,当A=1时,表明该材料不具有各向异性。通过计算得到A=0.55,表明立方BaBiO3具有各向异性。另外,根据杨氏模量E与弹性柔度之间的关系,立方结构的BaBiO3的各向异性可以由E检测。立方BaBiO3的杨氏模量E的方向依赖性如图7所示。从图7可看出,立方BaBiO3具有各向异性。

图7 立方BaBiO3杨氏模量(GPa)的方向依赖性
3 结束语
利用第一性原理对BaBiO3及使用Ta替代Bi在不同浓度掺杂下BaBiO3的电子结构和力学性能进行了计算分析。计算结果表明:1)经过结构优化后,晶格常数与实验值吻合;2) BaBiO3的面心立方结构由于DFT方法对带隙的低估与混合价态Bi原子电荷歧化,使得无法观察到带隙;3)对于Ta掺杂浓度分别为12.5%、25%、37.5%、50%四种情况,仅在50%浓度下,表现出了明显的带隙。4)通过BaBiO3力学性质的计算,表明该材料在立方结构下具有良好的延展性与韧性。
