菱镁矿和白云石的表面性质及浮游性的密度泛函理论研究
2020-12-17魏德洲高淑玲崔宝玉周世杰沈岩柏
韩 聪,魏德洲,高淑玲,崔宝玉,周世杰,沈岩柏
(东北大学资源与土木工程学院,辽宁 沈阳 110819)
镁是一种重要的金属资源,被广泛应用于材料、冶金、化工等领域,其氧化产物是重要的耐火材料之一[1]。菱镁矿是含量最丰富的镁矿资源,是镁的主要来源。我国菱镁矿资源储量位居世界第1位,但经过长时间的开采,高品位的菱镁矿资源越来越少,为保证菱镁矿资源的利用率及解决高品位菱镁矿短缺的问题,低品位菱镁矿的选矿工作势在必行[2-3]。浮选法是处理低品位菱镁矿矿石的主要方法,菱镁矿与白云石的高效分离是菱镁矿浮选技术的难点之一[4-6]。浮选是一个界面过程,使用浮选药剂对矿物表面的润湿性进行调节是浮选的核心问题。浮选药剂分子的吸附和电子转移都是在矿物界面发生和完成的,矿物的表面结构和性质则是浮选界面的基础。众多学者通过Zeta电位分析、表面形貌分析、能谱分析、吸附量测定、计算机模拟等方法[7-11],对菱镁矿和白云石在不同条件下的表面特性进行了深入的研究,获得了可靠的成果。但是,这些研究成果更多的是在宏观层次下对矿物表面性质的定性分析和解释,虽然已有部分学者从微观的原子层面对浮选过程中矿物的表面特性进行了研究,但这些研究主要集中在分析浮选药剂与矿物之间的相互作用,对矿物表面性质的研究还需进一步丰富。因此,从原子水平上系统地了解菱镁矿和白云石的表面特性及表面电子结构,对深入理解这两种矿物在不同条件下的浮选行为、浮选药剂与矿物之间的相互作用以及菱镁矿矿石的浮选分离技术都具有重要意义。
本文以菱镁矿和白云石为研究对象,以试验确定的矿物晶格常数为基础,采用基于密度泛函理论第一性原理的量子力学计算方法,对两种矿物的表面结构和性质进行研究,可以从原子水平上为两种矿物的浮选分离技术研究提供参考与指导。
1 研究方法
1.1 计算方法及主要参数设置
依据文献测定的菱镁矿晶格常数,a=b=4.636 Å,c=15.026 Å,α=β=90°,γ=120°[12];白云石的晶格常数:a=b=4.806 4 Å,c=16.006 Å,α=β=90°,γ=120°[13],在Materials Studio软件中分别构建两种矿物的初始晶胞模型,使用基于密度泛函理论第一性原理的量子力学程序CASTEP(Cambridge sequential total energy package)对矿物的晶体结构和表面结构进行优化。在此基础上,计算矿物表面的电子结构和相关性质。
计算中使用Ultrasoft赝势处理平面波,使用Pulay Density方法处理电子自洽,收敛精度为1×10-6eV/atom。矿物晶体几何结构优化和性质计算时体系能量、原子间作用力、原子位移以及压力的收敛精度分别设置为:2×10-5eV/atom、0.05 eV/Å、0.002 Å和0.1 GPa[14]。
1.2 表面模型构建
通过对比菱镁矿和白云石两种矿物晶格常数的计算值与实验值之间的误差,确定计算参数。通过对比菱镁矿和白云石表面结构模型的表面能,确定矿物表面结构模型。
矿物表面结构模型的表面能计算见式(1)[15]。

(1)
式中:Esurface为表面能;Nslab和Nbulk分别为矿物表面结构模型和体相模型中所包含的原子数量;Eslab和Ebulk分别为矿物表面结构模型和体相模型的总能量;A为矿物表面结构模型的表面积。
2 结果与讨论
2.1 菱镁矿和白云石晶体结构优化
2.1.1 交换-相关泛函对矿物晶格常数的影响
在截断能为310 eV,布里渊区K点取样密度为3×3×2时,采用不同交换-相关泛函计算了菱镁矿和白云石的晶格常数,结果见表1。其中,“晶格常数差”为晶格常数a、c计算值与实验值的误差之和。
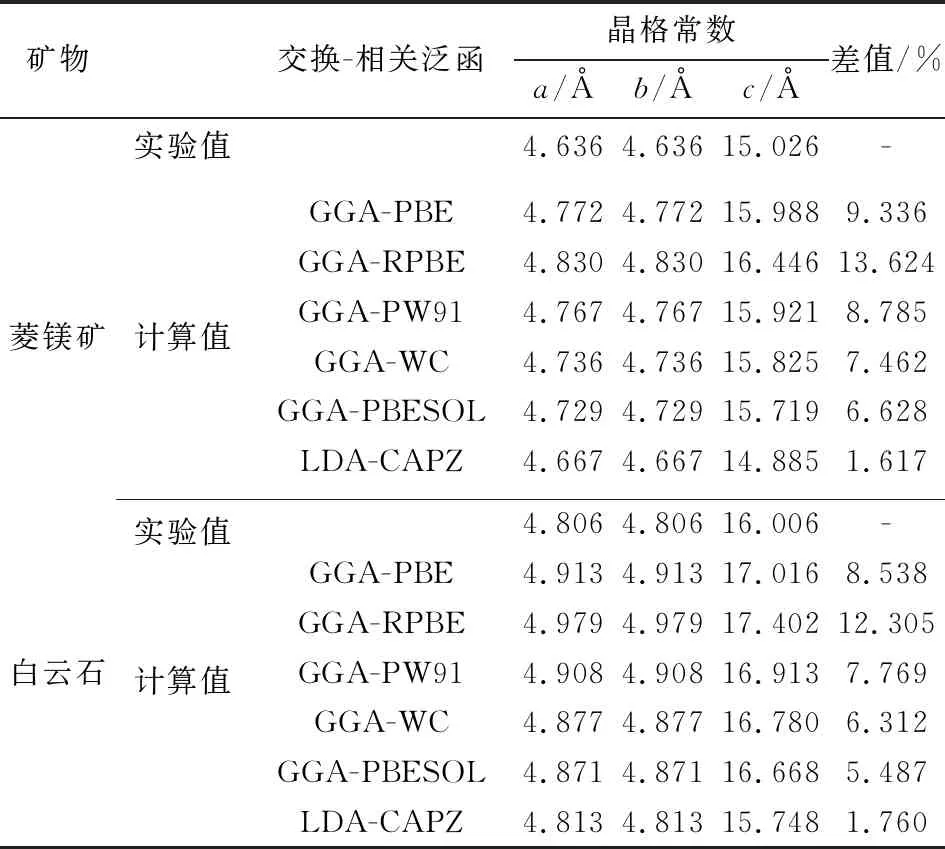
表1 不同交换-相关泛函条件下菱镁矿和白云石晶格常数的实验值与计算值对比Table 1 Comparison of lattice parameters for magnesite anddolomite lattices obtained from different DFT methodswith experimental values
由表1的计算结果可知,使用广义梯度近似(GGA)及与其关联的泛函计算得到的矿物晶格常数,计算值与实验值的误差均大于使用局域密度近似(LDA)及与其关联的泛函计算所得的误差。使用LDA+CA-PZ泛函时,菱镁矿和白云石晶格常数的计算值与实验值的误差最小,分别为1.617%和1.760%。
2.1.2 布里渊区K点密度对矿物晶格常数的影响
使用LDA近似及与其关联的CA-PZ泛函,截断能为310 eV的条件下,考察了布里渊区K点密度对菱镁矿和白云石晶格常数的影响,结果如图1所示。
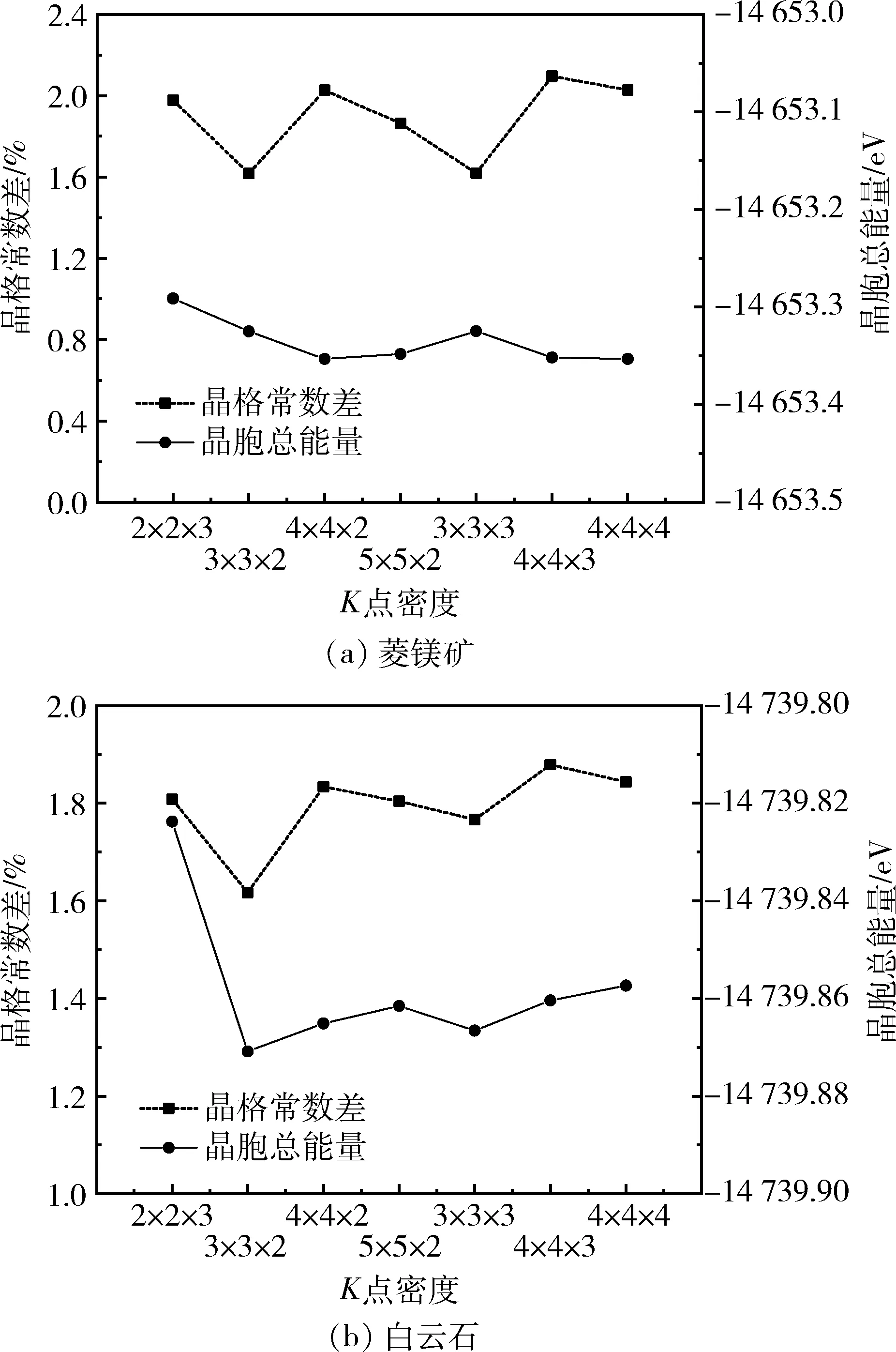
图1 截断能为310 eV时菱镁矿和白云石的晶胞总能量和晶格常数差与K点密度的关系曲线Fig.1 Total energies and lattice parameters differencesas a function of the K-points set mesh with the kineticcutoff energy of 310 eV for magnesite and dolomite
由图1(a)可以看出,菱镁矿的晶格常数差与K点密度关系曲线呈现波动状态,当K点密度为3×3×2和3×3×3时,晶格常数差较小且数值接近,考虑到增大K点取样密度会显著提高计算的运算量,固选取K点密度3×3×2计算菱镁矿晶体结构性质。由图1(b)可知,当K点密度为3×3×2时,白云石的晶格常数差最小。
2.1.3 平面波截断能对矿物晶格常数的影响
使用LDA近似及与其关联的CA-PZ泛函,布里渊区K点密度为3×3×2的条件下,考察了平面波截断能对菱镁矿和白云石晶格常数的影响,结果如图2所示。
由图2(a)可知,平面波截断能增大,菱镁矿的晶格常数差整体上呈现增大的变化趋势,当截断能为300 eV时,晶格常数差最小;由图2(b)可知,当平面波截断能为300 eV时,白云石的晶格常数差最小。
由以上结果可知,当使用LDA+CA-PZ交换-关联泛函、布里渊区K点密度3×3×2、平面波截断能300 eV时,菱镁矿和白云石晶格常数的计算值与实验值最为接近,计算结果见表2。
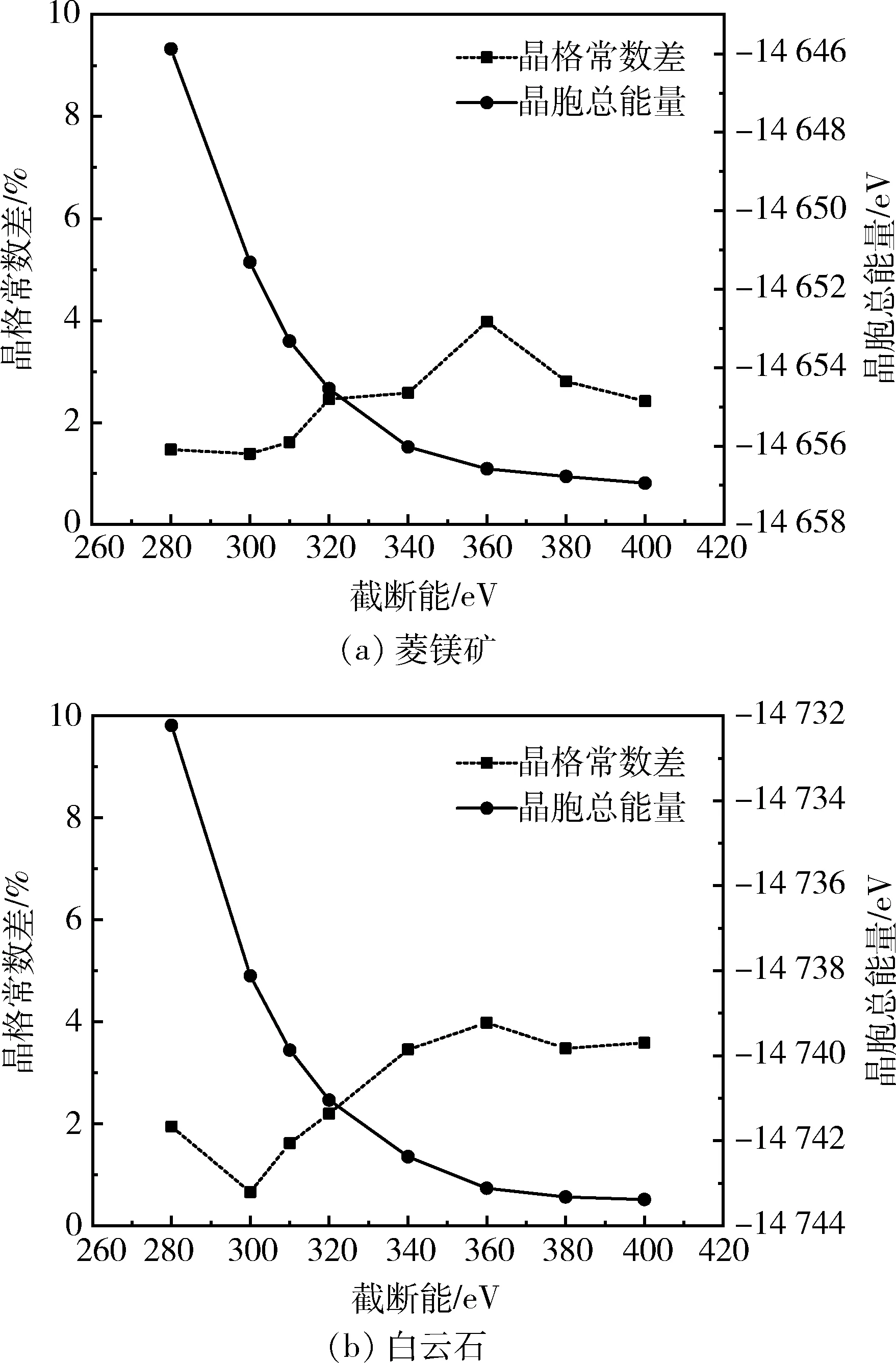
图2 K点密度为3×3×2时菱镁矿和白云石的晶胞总能量和晶格常数差与截断能的关系曲线Fig.2 Total energies and lattice parameters differencesas a function of kinetic cutoff energy with the K-pointmesh of 3×3×2 for magnesite and dolomite
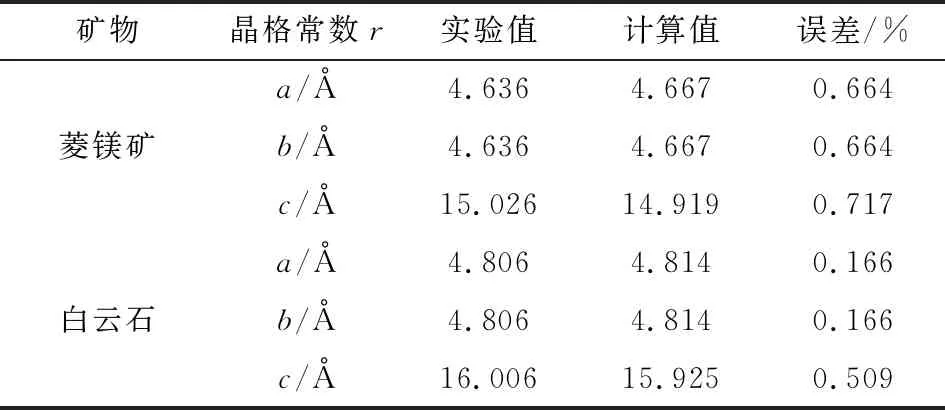
表2 菱镁矿和白云石晶格常数的计算值和实验值Table 2 Simulation and experimental lattice parametersof magnesite and dolomite crystal
由表2可知,菱镁矿晶格常数a、c的计算值与实验值的误差分别为0.664%和0.717%;白云石晶格常数a、c的计算值与实验值的误差分别为0.166%和0.509%,表明所选择的计算参数是合理的。
2.2 菱镁矿和白云石的表面结构与浮游性的关系
(101)面是菱镁矿和白云石的解理面。菱镁矿和白云石的晶体破裂时,(101)面是最容易形成的新表面,也是浮选中水分子、浮选药剂分子或离子最可能吸附的表面。矿物晶体沿某一晶面破裂形成新表面时,位于新生成表面上的原子将处于不均衡力场中并受力场作用进行弛豫,以改变表面结构,降低表面自由能,建立新的平衡状态。新形成表面的性质,如电荷分布、表面原子化学键的键性等将决定矿物表面的润湿性。
CASEP软件是在三维周期性条件下进行运算的,因此矿物表面结构模型表层原子的弛豫不仅受到来自矿物晶体内部原子作用力的影响,同时还受到表面间距的影响。为了考察表面原子弛豫对矿物表面结构的影响,计算了不同层面深度和真空层厚度的矿物层面模型的表面能,计算结果见表3;表面原子弛豫前后的矿物表面结构如图3所示。
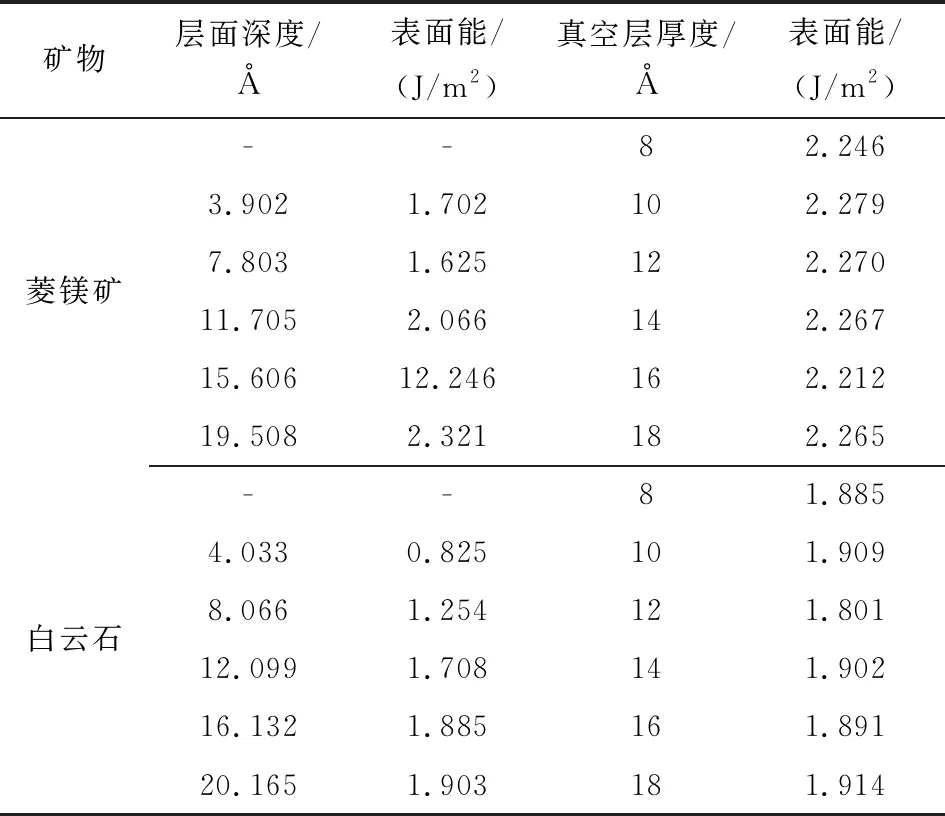
表3 不同层面深度和真空层厚度的菱镁矿和白云石晶体(101)面层面模型的表面能Table 3 Surface energies of magnesite (110) plane anddolomite (101) plane in function of slab depthand vacuum thickness
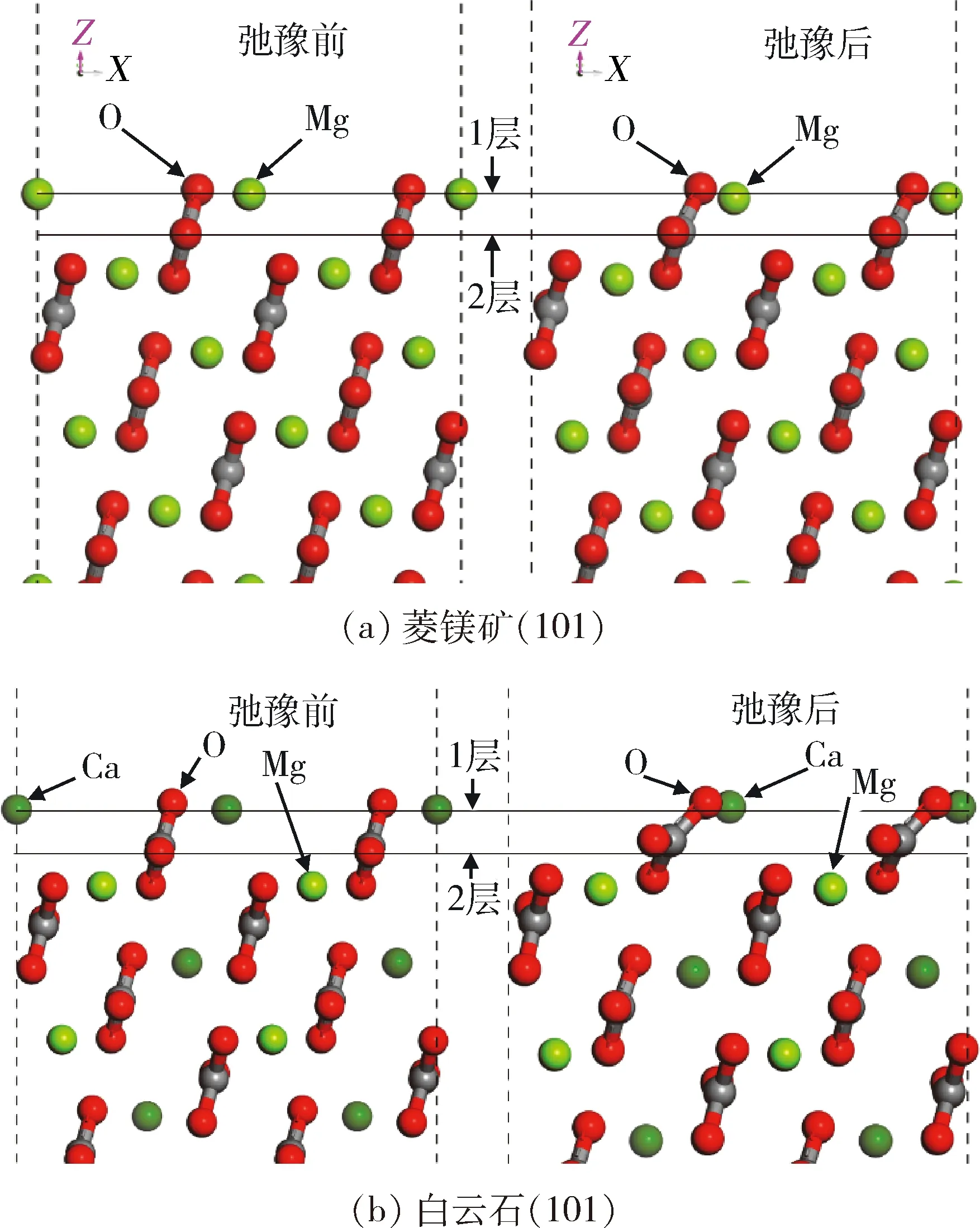
图3 菱镁矿(101)表面和白云石(101)表面的原子弛豫前后的矿物表面结构Fig.3 Structure of magnesite(101) plane slab modeland dolomite(101) plane slab model before andafter surface atoms relaxation
由表3可知,白云石(101)面的表面能小于菱镁矿(101)面的表面能;当菱镁矿层面深度达到15.606 Å(包含8层Mg-CO3单元)、白云石层面深度达到16.132 Å(包含4层Ca(Mg)[CO3]2单元)后,模型的表面能趋于稳定,其变化值在0.02 J/m2内;10 Å的真空层厚度可以避免由于三维周期性条件所导致的沿Z轴方向上真空层两侧表面之间的静电作用。
从图3可以看出,表面重构后两种矿物的表面原子均发生弛豫;白云石表面第1层氧原子的弛豫比菱镁矿第1层氧原子的弛豫明显;白云石表面第2层氧原子沿X轴的弛豫比菱镁矿第2层氧原子沿X轴的弛豫明显。
矿物表面晶格离子在矿浆中会溶解进入液相,当溶解达到饱和平衡时,矿物表面晶格离子进入液相的数目与同一时间内液相进入矿物晶格的离子数目相同。由表3和图3可知,白云石(101)面的表面能小于菱镁矿(101)面的表面能,并且两种矿物(101)面的表面结构相似,结合热力学知识可知,当液相中的Ca2+、Mg2+离子重新进入矿物晶格时,菱镁矿将形成类似白云石的表面结构,即菱镁矿的表面结构可以自发的向白云石的表面结构转化[7-8],使两种矿物的表面性质趋同,导致浮选分离的难度增大。
2.3 菱镁矿和白云石表面原子的电子结构与浮游性的关系
浮选药剂在矿物表面的吸附,是浮选研究最重要的内容之一。浮选药剂在矿物表面化学吸附的强弱,一方面取决于浮选药剂自身的性质;另一方面则由矿物表面原子的电子结构决定。分析矿物表面原子的电子结构,即可判断矿物表面原子的反应性[16-17]。菱镁矿(101)面和白云石(101)面表面原子的电子态密度计算结果如图4和图5所示。
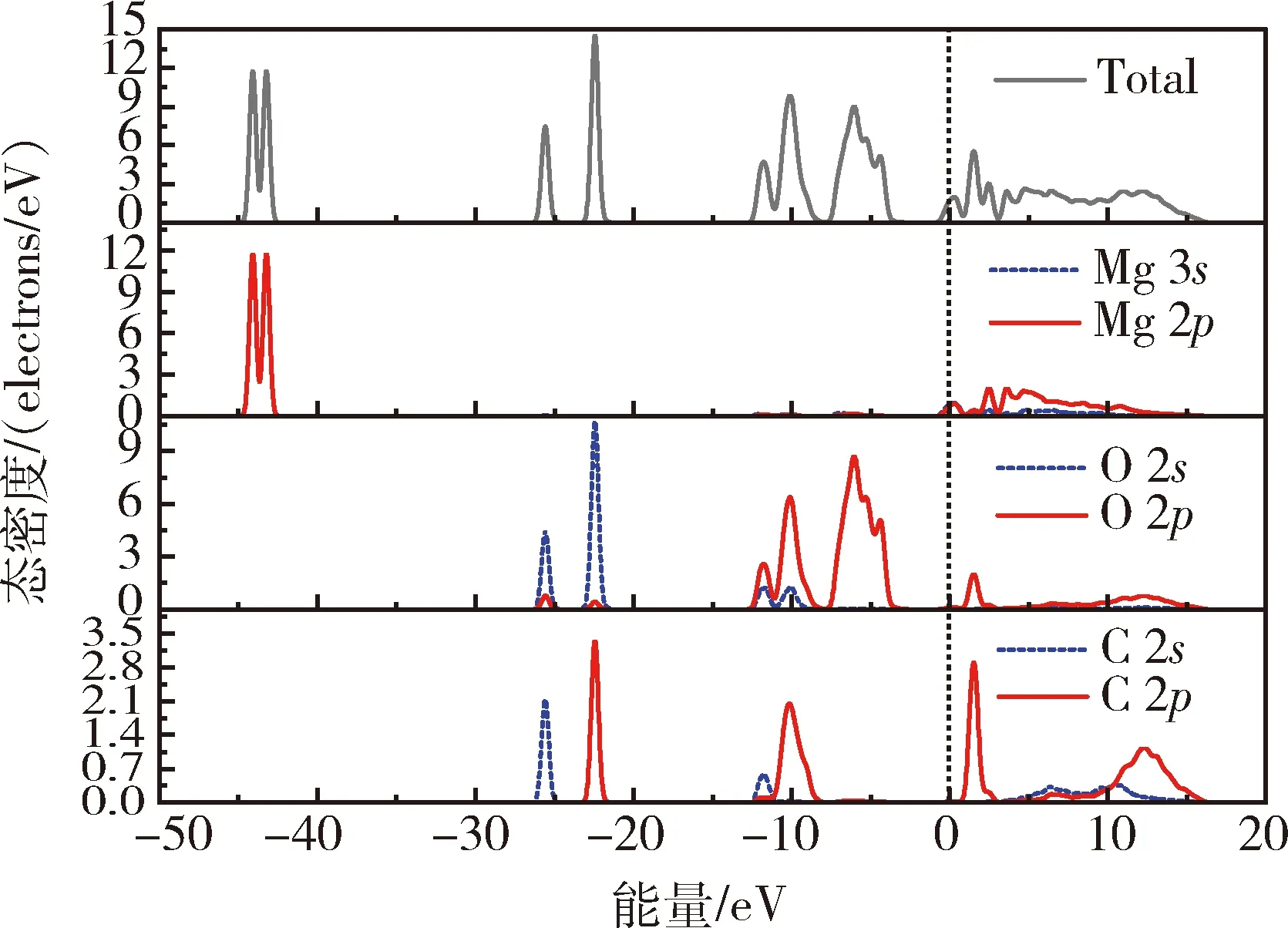
图4 菱镁矿(101)表面原子的总态密度和分波态密度Fig.4 Total and partial density of state of the atomsof the magnesite(101) surface
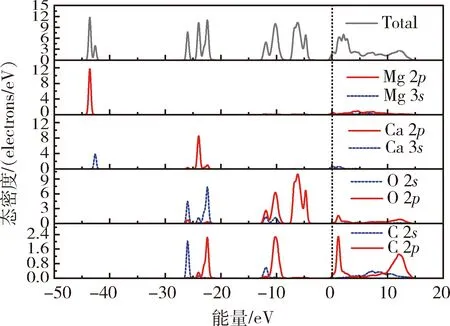
图5 白云石(101)表面原子的总态密度和分波态密度Fig.5 Total and partial density of state of the atomsof the dolomite(101) surface
由图4可知,菱镁矿的导带主要由Mg的3s轨道和2p轨道、O的2p轨道以及C的2s轨道和2p轨道贡献,上价带(-15.0~0 eV)主要由O的2s轨道和2p轨道以及C的2s轨道和2p轨道贡献,下价带(-30.0~-15.0 eV)主要由O的2s轨道以及C的2s轨道和2p轨道贡献,价带底由Mg的2p轨道贡献;在上价带中,O的2p轨道电子和C的2p轨道电子的态密度呈现尖峰态,说明位于2p轨道中的电子的有效质量较大、电子局域程度较高,轨道的扩展性较小,在化学反应中较难参与成键;Mg的3s轨道和2p轨道电子的能带跨越费米能级,表明位于Mg原子3s轨道和2p轨道内的电子能够较容易的从价带进入导带,说明在化学反应中Mg的3s轨道和2p轨道内的电子容易失去,即Mg原子是浮选药剂在菱镁矿表面化学吸附的活性位点。
由图5可知,白云石的导带主要由Mg的2p轨道、Ca的3s轨道、O的2p轨道以及C的2s轨道和2p轨道贡献,上价带(-15.0~0 eV)主要由O的2p轨道和C的2p轨道贡献,下价带(-30.0~-15.0 eV)主要由Ca的2p轨道、O的2s轨道以及C的2s轨道和2p轨道贡献,价带底由Mg的2p轨道和Ca的3s轨道贡献;在上价带中,O的2p轨道电子和C的2p轨道电子的态密度呈现尖峰态,电子局域性较强,在化学反应中较难参与成键;Ca的3s轨道电子和Mg的2p轨道及2s轨道电子的能带跨越费米能级,表明位于两种原子3s轨道和2p轨道内的电子能够容易的从价带进入导带,在化学反应中具有较大的活性。与Mg的3s、2p轨道电子在费米能及附近的态密度相比,Ca的3s轨道电子的峰更宽,说明在化学反应中Ca的3s轨道电子比Mg的3s和2p轨道电子的离域性强,即白云石表面的Ca原子比Mg原子的活性更强。
2.4 矿物表面不饱和化学键键性与浮游性的关系
矿物晶体经破碎后,矿物晶体表层原子朝向晶体内部的一面,与内层之间有平衡饱和键能,朝向晶体外部的键能没有得到饱和(或补偿),矿物表面这种未饱和的键能决定了他们的可浮性。矿物的表面键能按强弱可分为较强的离子键和较弱的分子键两大类,离子键具有较强的极性或化学活性,对极性的水分子有较大的吸引力,键的离子性越强,对水分子的吸引力越大,矿物表面越容易被水润湿,矿物的天然可浮性越差[18]。
通过对原子之间电荷密度的分析,可以研究原子间的成键。当成键的两个原子或官能团间最低电荷密度与背景电荷密度相等时,主要是离子键作用[19]。菱镁矿和白云石表面层原子的Mulliken电荷及其密度分布如图6所示。
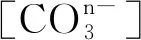
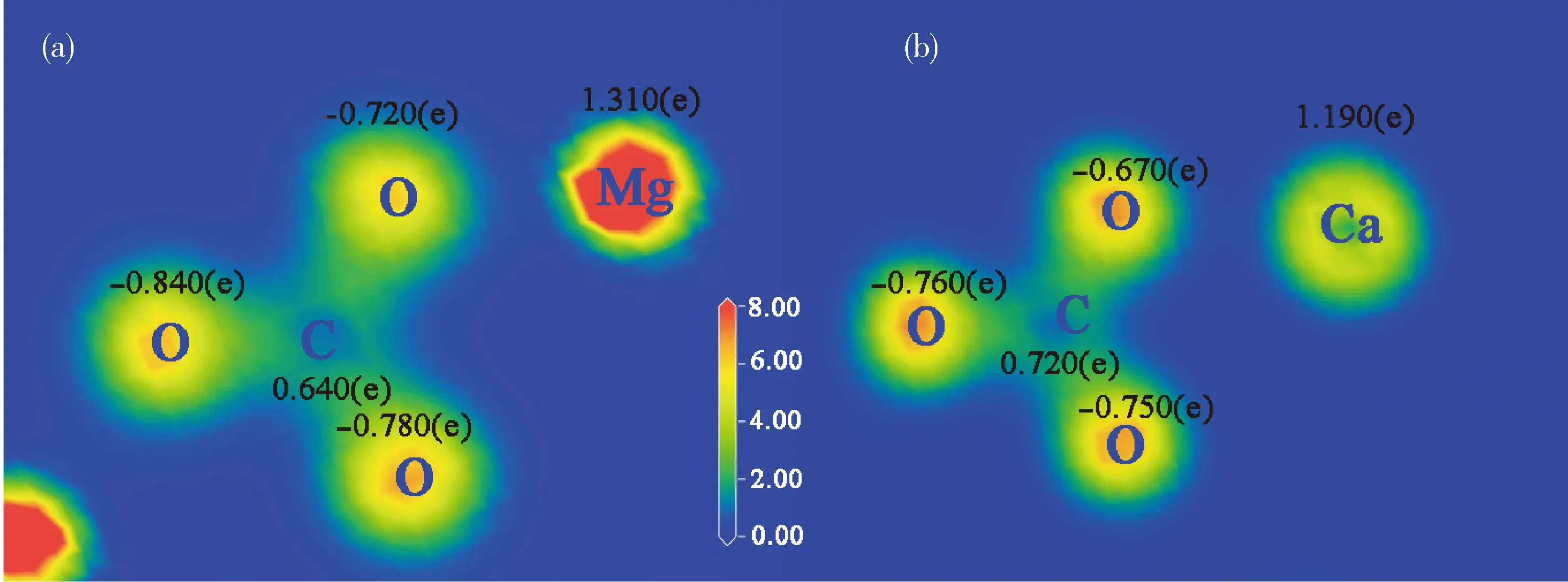
图6 菱镁矿(101)表面和白云石(101)表面原子的Mulliken电荷及密度分布Fig.6 Electron density and Mulliken charge of the surface atoms of the magnesite (101) surfaceand dolomite (101) surface
3 结 论
1) 使用基于密度泛函理论第一性原理的CASTEP软件研究菱镁矿和白云石晶体性质时,局域密度近似方法(LDA)相比广义梯度近似方法(GGA)可以更精确的描述矿物晶体的核外电子;采用LDA-CAPZ泛函、布里渊区K点密度为3×3×2、平面波截断能为300 eV时,菱镁矿和白云石晶格常数的计算值与试验值的误差最小。 决定菱镁矿和白云石解理面表面性质的原子主要位于距离菱镁矿表面15.606 Å(包含8层Mg-CO3单元)和白云石表面16.132 Å(包含4层Ca(Mg)[CO3]2单元)的范围内,当表面厚度分别大于这两个值后,两种矿物晶体表面的表面能趋于稳定。
2) 菱镁矿和白云石的表面重构达到热力学稳定状态时,菱镁矿的表面能大于白云石的表面能;矿浆中菱镁矿的表面可以自发的向白云石的表面转化,使两种矿物的表面性质趋同。
3) 菱镁矿和白云石表面的金属原子是浮选药剂化学吸附的活性位点;白云石表面的Ca原子比Mg原子的活性更强。 两种矿物表面的不饱和化学键均为离子键,对极性的水分子有较大的吸引力。
