齐墩果酸衍生物的合成及其与MEK靶点分子对接研究
2020-12-11张蓬勃宋艳玲
张蓬勃, 宋艳玲
(沈阳化工大学 制药与生物工程学院, 辽宁 沈阳 110142)
齐墩果酸(oleanolic acid,OA,图1)是一种五环三萜类化合物,具有显著的抗肿瘤、抗炎、抗氧化和抗血小板聚集等药理活性[1-2].但齐墩果酸的水溶性非常差,生物利用度低,大剂量服用会造成胆囊水肿,限制了其临床应用[3].含氮化合物广泛存在于自然界,许多有机含氮化合物具有显著的生物活性.文献报道三萜类化合物中引入氮原子能增加化合物的抗肿瘤活性,且含氮化合物与酸易生成盐,溶解性明显增加[4].席夫碱活性片段具有特殊的生理活性,多种含席夫碱的化合物具有抗肿瘤、抗菌和抑菌等药用活性[5-6].本文通过将席夫碱活性片段引入至OA-3位,设计并合成化合物Ⅰ1~Ⅰ4,通过席夫碱还原进一步设计并合成化合物Ⅱ1~Ⅱ4,以期提高目标化合物的生物活性和生物利用度.
近年来计算机辅助药物设计(computer aided drug design,CADD)在新药研发过程中发挥着重要作用.分子对接研究是将配体分子放置于受体分子的活性位点,利用化合物与受体蛋白的三维结构进行对接,预测化合物的生物活性.ERK(extracellular regulated kinase)是丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)家族的重要成员,其信号转导通路(Ras-Raf-MEK-ERK)调控异常会导致多种癌细胞增生、扩散并转移[7].MEK作为ERK途径中的重要靶点,已成为抗肿瘤药物的研究热点.本文通过计算机辅助设计的方法研究OA衍生物与MEK分子对接,预测目标化合物抑制MEK的活性.

图1 齐墩果酸的立体化学结构
1 实验部分
1.1 仪器与试剂
Bruker ARX-600型核磁共振仪,美国Bruker公司;Büchi B-540熔点测定仪,瑞士Büchi公司;薄层色谱(GF-254),柱色谱硅胶(200~300目),青岛海洋化工有限公司;齐墩果酸(质量分数为98 %),陕西慈缘生物技术有限公司;芳香醛类药品,国药集团化学试剂有限公司.其他溶剂和药品均为分析纯.
1.2 合成实验
OA衍生物的合成路线见图2.
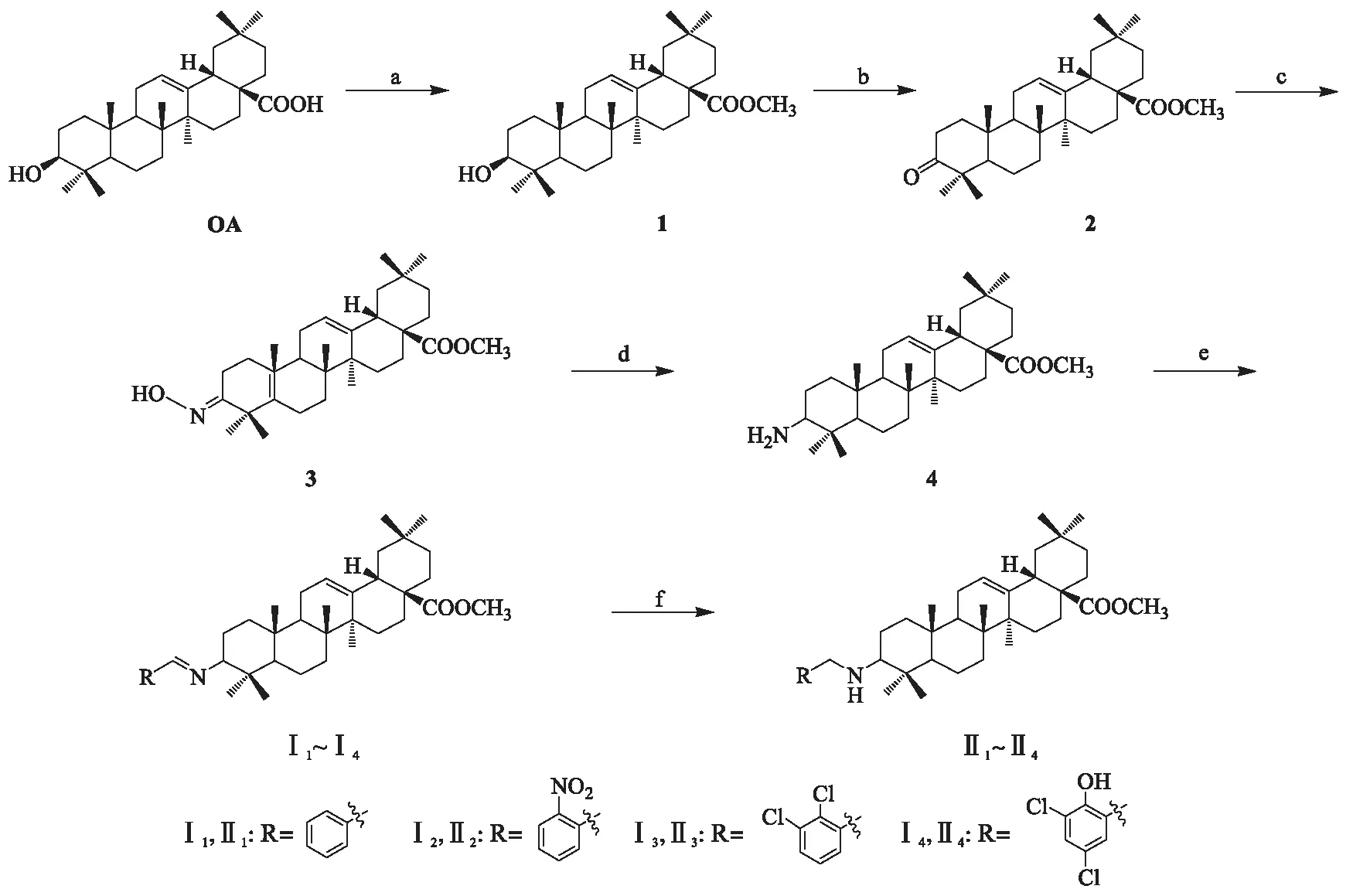
试剂和反应条件:(a) K2CO3,CH3I,DMF,室温;(b) 琼斯试剂,丙酮,异丙醇,0 ℃;(c) NH2OH·HCl,Py,50 ℃;(d) NaBH3CN,TiCl3,CH3COONH4,CH3OH,室温;(e) 芳香醛,乙醇,80 ℃;(f) NaBH3CN,乙酸,室温
1.2.1 齐墩果酸甲酯(1)的制备
将OA(5.00 g,10.94 mmol)和无水K2CO3(3.78 g,27.35 mmol)溶于50 mL干燥的N,N-二甲基甲酰胺中,搅拌下缓慢加入碘甲烷(1.37 mL,21.89 mmol),室温下继续搅拌反应6 h,TLC监测反应.反应结束后加水稀释,乙酸乙酯萃取,饱和NaCl溶液洗涤,水洗,合并有机相.无水硫酸钠干燥,抽滤,减压蒸干溶剂,得白色固体.干燥过夜,甲醇重结晶得白色针状晶体化合物14.40 g,产率85 %,m.p.196~198.8 ℃(文献m.p.197~198.2 ℃)[8].
1.2.2 3-羰基-齐墩果酸甲酯(2)的制备
将齐墩果酸甲酯(1)(4.40 g,9.35 mmol)溶于100 mL丙酮中,冰浴下缓慢滴加Jones试剂(11.5 mL),直至反应液橘红色不再消失,滴毕,室温下反应1~2 h,TLC监测反应.反应完毕,加入130 mL异丙醇淬灭.反应结束后,减压蒸除部分溶剂,乙酸乙酯萃取,饱和NaCl溶液洗涤,水洗,合并有机相.无水硫酸钠干燥,抽滤,减压蒸干,得淡黄绿色固体.干燥,甲醇重结晶得白色晶体化合物24.15 g,产率95 %,m.p.186~187 ℃(文献m.p.185~186 ℃)[9].
1.2.3 3-羟肟基-齐墩果酸甲酯(3)的制备
将3-羰基-齐墩果酸甲酯(2)(4.15 g,8.87 mmol)溶于50 mL吡啶中,加入盐酸羟胺(2.76 g,39.74 mmol),50 ℃下反应2 h,TLC监测反应.反应结束后加入适量二氯甲烷稀释,用质量分数为10 %的盐酸洗涤3次,饱和NaCl溶液洗涤,水洗,合并有机相.无水硫酸钠干燥,抽滤,减压蒸干,得白色固体化合物33.24 g,产率75.5 %,m.p.244~246 ℃(文献m.p.247~249 ℃)[9].
1.2.4 3-氨基-齐墩果酸甲酯(4)的制备
将3-羟肟基-齐墩果酸甲酯(3)(4.88 g,10.10 mmol)溶于100 mL甲醇中,加入乙酸铵(9.04 g,117.30 mmol),再加入NaBH3CN(7.08 g,112.70 mmol),充分搅拌溶解,冰浴下缓慢滴加质量分数为15 %的三氯化钛溶液(29.33 mL,34.22 mmol),室温下反应12 h,TLC监测反应.反应结束后,蒸除部分溶剂,用2 mol/L的NaOH溶液调pH至10,二氯甲烷萃取,蒸馏水洗涤,合并有机相.无水硫酸钠干燥,抽滤,减压蒸干,硅胶柱层析分离(200~300目),洗脱剂为甲醇/二氯甲烷,得白色固体化合物43.78 g,产率80 %,m.p.221.3~223.7 ℃.ESI-MS,m/z:470.8[M+H]+;1H-NMR(600MHz,CDCl3),δ:5.29(1H,s,H-12),3.64(3H,s,28-COOCH3),2.87(1H,d,J=13.6 Hz,H-18),2.51(1H,d,J=9.7 Hz,H-2),1.14(3H,s,CH3),1.02(3H,s,CH3),0.94(3H,s,CH3),0.91(6H,s,2CH3),0.81(3H,s,CH3),0.74(3H,s,CH3).
1.2.5 化合物Ⅰ1~Ⅰ4的制备
1.2.5.1 化合物Ⅰ1的制备
将3-氨基-齐墩果酸甲酯(0.15 g,0.32 mmol)溶于10 mL无水乙醇中,苯甲醛(0.041 g,0.384 mmol)溶于5 mL无水乙醇中,随后将芳香醛乙醇溶液逐滴加入反应体系中,80 ℃条件下回流反应4~6 h.溶液变浑浊,有固体析出,抽滤,甲醇重结晶得白色固体产物化合物Ⅰ10.12 g,m.p.192~194 ℃,产率63 %.1H-NMR(600 MHz,CDCl3),δ:8.26(s,1H),7.74~7.72(m,2H),7.41~7.37(m,3H),5.31(s,1H),3.63(s,3H),1.16(s,3H),1.01(s,3H),0.98(s,3H),0.93(s,3H),0.90(s,3H),0.78(s,3H),0.76(s,3H).
1.2.5.2 化合物Ⅰ2的制备
实验步骤同化合物Ⅰ1.3-氨基-齐墩果酸甲酯(0.15 g,0.32 mmol)与邻硝基苯甲醛(0.058 g,0.384 mmol)反应,得到棕色固体产物化合物Ⅰ20.118 g,m.p.193~196 ℃,产率57 %.1H-NMR(600 MHz,CDCl3),δ:8.66(s,1H),8.01~7.98(m,2H),7.64(d,J=7.6 Hz,1H),7.53(t,J=8.0 Hz,1H),5.31(s,1H),3.63(s,3H),1.16(s,3H),1.01(s,3H),0.98(s,3H),0.93(s,3H),0.91(s,3H),0.82(s,3H),0.76(s,3H).
1.2.5.3 化合物Ⅰ3的制备
实验步骤同化合物Ⅰ1.3-氨基-齐墩果酸甲酯(0.15 g,0.32 mmol)与3,4-二氯苯甲醛(0.068 g,0.384 mmol)反应,得到白色固体产物化合物Ⅰ30.116 g,m.p.192~195 ℃,产率53 %.1H-NMR(600 MHz,CDCl3),δ:8.17(s,1H),7.83(d,J=1.9 Hz,1H),7.54~7.52(m,1H),7.46(d,J=8.3 Hz,1H),5.31(s,1H),3.63(s,3H),1.16(s,3H),1.00(s,3H),0.96(s,3H),0.93(s,3H),0.90(s,3H),0.76(s,6H).
1.2.5.4 化合物Ⅰ4的制备
实验步骤同化合物Ⅰ1.3-氨基-齐墩果酸甲酯(0.15 g,0.32 mmol)与3,5-二氯水杨醛(0.074 g,0.384 mmol)反应,得到黄色固体产物化合物Ⅰ40.161 g,m.p.195~196 ℃,产率72 %.1H-NMR(600 MHz,CDCl3),δ:15.04(s,1H),8.18(s,1H),7.41(d,J=2.5 Hz,1H),7.13(d,J=2.6 Hz,1H),5.30(s,1H),1.15(s,3H),0.98(s,3H),0.96(s,3H),0.93(s,3H),0.90(s,3H),0.85(s,3H),0.75(s,3H).
1.2.6 化合物Ⅱ1~Ⅱ4的制备
1.2.6.1 化合物Ⅱ1的制备
将3-氨基-齐墩果酸甲酯(0.15 g,0.32 mmol)溶于10 mL无水乙醇中,将苯甲醛(0.041 g,0.384 mmol)用5 mL无水乙醇溶解后逐滴加到反应瓶中,80 ℃条件下回流反应4~6 h,冷却至室温后,加入NaBH3CN(0.06 g,0.96 mmol),室温条件下反应2 h,TLC监测反应.反应结束后加入5 mL冰醋酸进行淬灭,二氯甲烷萃取,饱和NaCl溶液洗涤3次,合并有机相.无水硫酸钠干燥,抽滤,减压悬蒸,硅胶柱层析分离(200~300目),洗脱剂为石油醚/乙酸乙酯,得到白色固体产物化合物Ⅱ10.08 g,m.p.187~190 ℃,产率42 %.1H-NMR(600 MHz,CDCl3),δ:7.35(d,J=7.1 Hz,2H),7.31(t,J=7.6 Hz,2H),7.25~7.21(m,2H),5.28(s,2H),3.95(d,J=13.2 Hz,1H),3.67(d,J=13.2 Hz,1H),3.62(s,3H),1.11(s,3H),0.97(s,3H),0.92(s,3H),0.90(s,6H),0.74(s,3H),0.72(s,3H).
1.2.6.2 化合物Ⅱ2的制备
实验步骤同化合物Ⅱ1.3-氨基-齐墩果酸甲酯(0.15 g,0.32 mmol)与邻硝基苯甲醛(0.058 g,0.384 mmol)反应,得到棕色固体产物化合物Ⅱ20.083g,m.p.182~186 ℃,产率40 %.1H-NMR(600 MHz,CDCl3),δ:7.89(d,J=8.1 Hz,1H),7.66(d,J=7.7 Hz,1H),7.55(t,J=7.5 Hz,1H),7.38(t,J=7.7 Hz,1H),5.28(s,1H),4.20(d,J=14.3 Hz,1H),3.91(d,J=14.3 Hz,1H),3.62(s,3H),1.12(s,3H),0.94(s,3H),0.93(s,3H),0.90(d,J=2.2 Hz,6H),0.74(s,3H),0.72(s,3H).
1.2.6.3 化合物Ⅱ3的制备
实验步骤同化合物Ⅱ1.3-氨基-齐墩果酸甲酯(0.15 g,0.32 mmol)与3,4-二氯苯甲醛(0.068 g,0.384 mmol)反应,得到白色固体产物化合物Ⅱ30.076 g,m.p.185~186 ℃,产率35 %.1H-NMR(600 MHz,DMSO-d6),δ:7.61(s,1H),7.54(d,J=8.2 Hz,1H),7.34(d,J=8.2 Hz,1H),5.18(s,1H),3.84(d,J=14.3 Hz,1H),3.61(d,J=14.5 Hz,1H),3.54(s,3H),1.07(s,3H),0.91(s,3H),0.87(d,J=2.7 Hz,6H),0.85(s,3H),0.69(s,3H),0.65(s,3H).
1.2.6.4 化合物Ⅱ4的制备
实验步骤同化合物Ⅱ1.3-氨基-齐墩果酸甲酯(0.15 g,0.32 mmol)与3,5-二氯水杨醛(0.074g,0.384 mmol)反应,得到黄色固体产物化合物Ⅱ40.10g,m.p.182~184 ℃,产率45 %.1H-NMR(600 MHz,DMSO-d6),δ:13.11(s,1H),7.29(d,J=2.6 Hz,1H),7.08(d,J=2.6 Hz,1H),5.19(s,1H),4.07(d,J=14.7 Hz,1H),3.85(d,J=14.6 Hz,1H),1.09(s,3H),0.99(s,3H),0.88(d,J=2.5 Hz,6H),0.86(s,3H),0.73(s,3H),0.66(s,3H).
1.3 化合物与MEK的分子模拟对接
从蛋白数据库(protein data bank,PDB)(http://www.rcsb.org/)中下载MEK靶蛋白结构(PDB编号:4MX5).在ChemDraw中以structure选项下的convert name to structure功能分别画出其结构图,并以MDL Molfile(*.mol)格式存储,使用Discovery studio 4.0软件导入上步建立的MDL Molfile(*.mol)格式文件,使用其convert功能,将MDL Molfile格式转换成为sdf-MDL MOL format(*.mol2)格式文件.利用计算机辅助药物设计软件Molegro Virtual Docker(MVD 6.0)对各分子对接的结合能进行测试,用Discovery studio 4.0分析对接模式.
2 结果与讨论
MVD软件能够通过函数打分的形式,预测并计算蛋白质与小分子配体的相互作用,结果以结合自由能(Escore=Einter+Eintra,kJ/mol)形式表达,Einter表示配体-蛋白质相互作用能量,Eintra表示配体内部能量.结合能绝对值越大,表示结合亲和力越强.分子对接的结合能结果见表1.
研究结果表明:化合物Ⅰ1~Ⅰ4和Ⅱ1~Ⅱ4与MEK靶蛋白(PDB编号:4MX5)的结合能均高于OA,且Ⅱ系列高于Ⅰ系列,表明席夫碱还原后的化合物与靶蛋白具有更高的结合能,其中化合物Ⅱ4的结合能高达-552.36 kJ/mol,超过MEK靶蛋白抑制剂曲美替尼的结合能-538.63 kJ/mol.曲美替尼是一种代表性的小分子MEK抑制剂[10],现通过分子模拟对接方法研究其与MEK靶蛋白(PDB编号:4MX5)的相互作用(图3).分子对接结果表明:曲美替尼可以和MEK蛋白氨基酸残基形成多个牢固的氢键,与靶蛋白产生较大结合力.化合物Ⅱ4与靶蛋白的分子模拟对接结果表明:Ⅱ4能较好地插入到靶点的活性口袋中,其结构中的酯基、芳香羟基通过氢键与靶蛋白氨基酸残基(SER:176,GLU:177,ARG:258,VAL:81)紧密结合;还原席夫碱亚甲基片段、芳香取代基团通过碳氢键、疏水键、π—σ 键和π—alkyl键等与靶蛋白多个氨基酸残基(CLU:177,SER:176,ARG:213,TYR:123,ILE:172)结合,如图4所示.
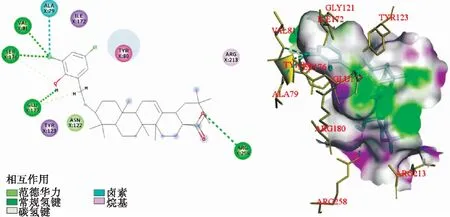
图4 化合物Ⅱ4与MEK靶蛋白(PDB编号:4MX5)分子对接模式
3 结 论
设计并合成了8个未见文献报道的OA衍生物,通过分子对接的方法模拟目标化合物和MEK靶蛋白大分子相互作用.研究结果表明:化合物Ⅱ4可通过氢键与MEK靶蛋白多个氨基酸残基(SER:176,GLU:177,ARG:258,VAL:81)结合,产生较高的结合能.可通过药理活性实验进一步研究其抑制MEK的活性.计算机辅助筛选方法可大大提高新药研发的速度,降低新药研发的成本,同时分子模拟对接模式的研究对药物作用机制研究具有重要意义.
