全基因组预测哈茨木霉的效应子
2020-12-10陈欣瑜董章勇
罗 梅,陈欣瑜,2,董章勇,2*
(1.仲恺农业工程学院 农业与生物学院,广东 广州 510225;2.仲恺农业工程学院植物健康创新研究院,广东 广州 510225)
【研究意义】哈茨木霉(Trichodermaharzianum)是木霉菌的一种,属半知菌类(Fungi imperfecti),丝孢纲(Hyphomycetes),丛梗孢目,丛梗孢科,是一种目前广泛应用的重要生防菌。该属广泛分布于全球,在土壤及有机质上均可发现[1]。哈茨木霉报道能够与长孺孢属(Helmisporiumspp.)、核盘菌属(Sclerotiniaspp.)、轮枝孢属(Verticilliumspp.)、黑星菌属(Venturiaspp.)、蜜环菌属(Armillariaspp.)、镰刀菌(Fusariumspp.)、腐霉菌(Pythiumspp.)、疫霉菌(Phytophthoraspp.)、丝核菌(Rhizoctoniaspp.)、炭疽菌(Colletotrichumspp.)等多种植物病原菌竞争生长空间和营养物质[2-4],且能分泌产生一些抑菌物质,具有重寄生作用,并可以诱导植物产生抗性[5-7]。因此,其菌剂被广泛应用在植物病害的防治上[6-7]。以色列的哈茨木霉T39和美国T22 菌株等均已经登记使用在多种植物病害的防治上。【前人研究进展】效应子(effector)也称效应因子或效应蛋白,广义上是指能够选择性地结合其他目标蛋白及调节其生物活性的一类小分子蛋白。随着对效应子的研究,有学者凝炼出其具体的分析指标:①含有N-端信号肽;②无跨膜结构域;③定位于细胞质;④序列具有特异性;⑤富含半胱氨酸;⑥重复元件多;⑦氨基酸残基数量大约在50~300 氨基酸[8-9]。哈茨木霉菌株的基因组测序的完成为高通量筛选效应子提供前提。许僖等[10]根据哈茨木霉T677菌株的基因组预测了其分泌蛋白,但并未进行效应子的分析。【本研究切入点】本研究在已公布的哈茨木霉CBS 226.95菌株全基因组信息的基础上,根据效应子的典型特征,利用生物信息学软件对其候选效应子进行了预测。【拟解决的关键问题】研究结果为哈茨木霉效应子的进一步筛选与验证提供了重要的理论依据,并为探究效应子在生防菌与病原菌互作过程中的作用奠定基础。
1 材料与方法
1.1 基因组信息
哈茨木霉CBS 226.95菌株的蛋白序列从NCBI上下载,具体网址为:ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/003/025/095/GCF_003025095.1_Triha_v1.0/GCF_003025095.1_Triha_v1.0_protein.faa.gz。该基因组共包含了14 065个蛋白序列(INSDC: MBGI00000000.1)。
1.2 分泌组蛋白预测
将14 065个蛋白序列先经过SignalP v4.1分析;随后将有信号肽的蛋白进一步用TMHMM v2.0进行分析,选取跨膜结构域小于2 的蛋白;进一步用TargetP v1.1进行分析,筛选定位在胞外的蛋白;然后用ProtComp v9.0进行分析亚细胞定位。
1.3 分泌组蛋白序列分析
将1.2分析结果进行序列长度统计分析。
1.4 分泌组蛋白的半胱氨酸含量和multiple tandem repeats分析
将1.2分析结果的蛋白序列进行半胱氨酸数量及multiple tandem repeats分析。半胱氨酸数量采用perl工具进行计算。将具有高半胱氨酸的 (≥6) 的蛋白序列进行multiple tandem repeats分析。multiple tandem repeats采用T-Reks (http://bioinfo.montp.cnrs.fr/?r= t-reks/)进行预测。
1.5 候选致病相关效应因子的确定
一般的效应子都是小分子蛋白,因此,将1.4获得的蛋白序列去除大于300个氨基酸的蛋白序列,剩余的为候选效应子。将获得的蛋白序列和PHI数据库(Pathogen-Host Interaction database,http://www.Phi-bas e.org/downloadLink.htm)进行比对分析,分析其可能的功能。
2 结果与分析
2.1 分泌组蛋白预测
将14 065个哈茨木霉蛋白序列用SignalP v4.1进行分析,共获得1288个蛋白具有信号肽;随后进一步用TMHMM v2.0进行跨膜结构分析,跨膜结构域小于2的共有1186个蛋白;将这1186个蛋白用TargetP v1.1进行定位分析,共得到1138个蛋白在胞外表达;再进一步用ProtComp v9.0进行分析,得到709个蛋白。这709个蛋白为哈茨木霉CBS226.95菌株的分泌蛋白,占总蛋白的5.04 %。
2.2 分泌组蛋白序列分析
将709个蛋白氨基酸残基序列特征进行分析,获得序列长度统计数据(perl程序)。小于200个氨基酸的有199个,占28 %;201~400个氨基酸的有275个序列,占39 %;401~600个氨基酸的有149个序列,占21 %;601~800个氨基酸的有57个序列,占8 %;801~1000个氨基酸的有23个序列,占3 %;大于1000个氨基酸残基的有6个序列,占1 %;没有大于2000个氨基酸残基的序列(图1)。
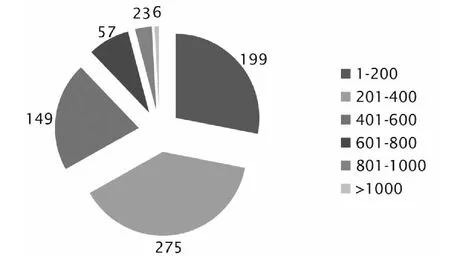
图1 709个哈茨木霉分泌蛋白的长度统计分析Fig.1 Sequence length statistics of 709 Trichoderma harzianum secretome genes
2.3 分泌组蛋白的cysteine含量和multiple tandem repeats分析
将709个蛋白序列进行cysteine含量及multiple tandem repeats分析。Cysteine含量采用perl工具进行计算。得到396个具有高cysteine含量的 (≥6) 的蛋白。其中6个半胱氨酸的有93个序列,占23.5 %,其次为8个半胱氨酸,有72个序列,占18.2 %;再次为7、10个,分别为45和47个,占11.4 %和11.9 %;其中1个序列(XP_024772708.1)拥有98个半胱氨酸位点(图2)。
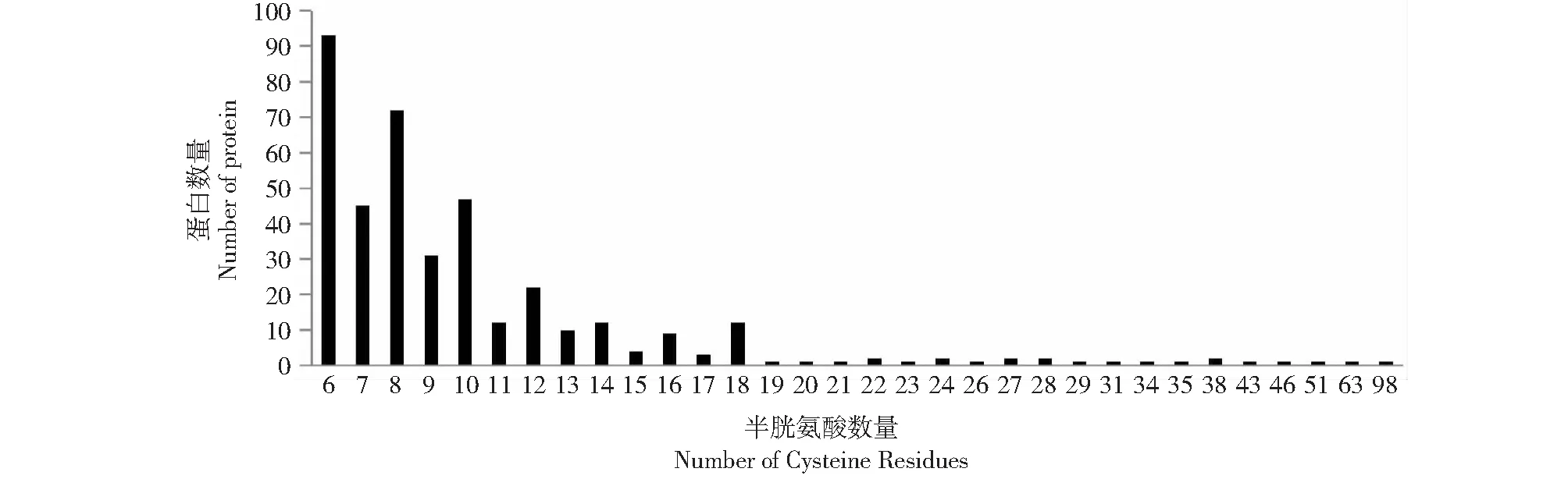
图2 396个哈茨木霉分泌蛋白的半胱氨酸残基数量分布Fig.2 Number of cysteine residues of the 396 Trichoderma harzianum secretome genes
将396个具有高cysteine含量的 (≥6) 的蛋白序列进行multiple tandem repeats分析。multiple tandem repeats采用T-Reks (http://bioinfo.montp.cnrs.fr/?r=t-reks/)进行预测。得到86个蛋白具有multiple tandem repeats (≥9)。
2.4 候选致病相关效应因子的确定
一般效应因子都是小分子蛋白,因此,将上述获得的86个基因序列进行长度筛选,去除大于300个氨基酸的蛋白序列。剩余24个基因序列,被认为是候选致病相关效应因子(表1)。其中有3个是碳水化合物结合模块家族蛋白(carbohydrate-binding module family protein),其余21个为假定蛋白。在PHI数据库比对结果显示,其中9个蛋白可能跟真菌的毒力相关(reduced virulence),3个为效应子(effector),2个蛋白是致死蛋白(lethal),6个为非致病相关(Unaffected pathogenicity),1个蛋白为功能未知,1个蛋白为混合型(可能为非致病相关或引起毒力减弱)。

表1 哈茨木霉候选效应因子

续表1 Continued table 1
3 结论与讨论
分泌的胞外蛋白除了作为效应子参与侵染寄主过程外,还可以作为毒力因子或者毒素改变寄主细胞的结构或者功能,或者作为无毒因子或者激发子引发寄主防卫反应[11-12]。本研究通过对哈茨木霉CBS 226.95菌株的14 065个蛋白进行分析,共分析得到709个分泌蛋白,占总蛋白的5.04 %。而许僖等[10]根据哈茨木霉T677菌株基因组的11 498个蛋白中共分析得到分泌蛋白503个,占总蛋白的4.37 %。不同的植物真菌所含分泌蛋白占的比列不同,多数半活体营养型和死体营养型真菌编码的分泌蛋白占总基因的比例高于活体营养型真菌[13]。本研究分析到的分泌蛋白的比例比许僖等[10]分析的略高,可能2个菌株来源不一样的差异引起的。
本研究基于哈茨木霉CBS 226.95菌株基因组共筛选获得24个效应子,其中有3个是碳水化合物结合模块家族蛋白(carbohydrate-binding module family protein,CBM),其余21个为假定蛋白。虽然通过PHI数据库对其功能进行了初步的注释,但是相应蛋白在哈茨木霉的具体功能还需要进一步进行确认。3个碳水化合物结合模块家族蛋白中有1个是CBM50 亚家族,2个是CBM1亚家族。CBM作为碳水化合物酶类的一大类,在真菌的作用机制上发挥了重要作用。其中CBM50亚家族属于CBM的一个亚家族,通常结合糖基水解酶家族(glycoside hydrolases,GH)发挥降解几丁质或者肽聚糖的作用(http://www.cazy.org/CBM50.html)。柳少燕等[14]分析表明拮抗菌中的CBM50相比病原菌来说可能发生了显著的扩增,且有部分拮抗菌中的CBM50蛋白单独聚为一支。本研究所分析获得的CBM50亚家族基因具有1个LysM结构域。LysM结构域最早是在芽孢杆菌噬菌体溶菌酶中被发现其通过降解N-乙酰胞壁酸和N-乙酰葡糖胺之间的糖苷键来降解细胞壁[15]。LysM结构域偏好出现在蛋白质的N端和C端,在中心位置出现较少[16]。本研究所分析的CBM50蛋白的LysM结构域出现在C端。由于拮抗菌与病原菌的细胞组分相似,拮抗菌需要解决如何识别“自我”与“非我”并避免自身受到影响的关键问题。LysM结构域作为一类古老而又普遍存在的结构域,对该类结构域的研究有助于揭示拮抗真菌、病原真菌及植物防御之间的复杂联系。Chen等[25]报道效应子LysM帮助逃避昆虫的免疫防御而致病昆虫。
随着基因组学以及生物信息学的快速发展,大数据分析成为现代生物学研究的重要研究手段之一[10, 17-19]。通过高通量的方法进行效应子的筛选,虽然可能不完全,但可以大大的缩小研究范围,找到研究的切入口。而本研究筛选到的潜在的效应子在哈茨木霉的拮抗作用以及在植物环境中的作用还需要进行进一步的功能研究才能得以验证。
