HPLC法测定甲磺酸培氟沙星胶囊的含量及有关物质
2020-12-09朱晓璐胡幸
朱晓璐,胡幸
(重庆市食品药品检验检测研究院,重庆 401121)
0 引言
甲磺酸培氟沙星是喹诺酮类第三代抗菌药物,主要作用于细菌细胞的DNA旋转酶,干扰细菌DNA的合成,使细菌死亡[1]。其合成工艺系由诺氟沙星与甲醛、甲酸进行甲基化反应生成培氟沙星,再与甲磺酸在乙醇溶液中进行成盐反应[2-3]。甲磺酸培氟沙星胶囊收载于《中国药典》2015年版,甲磺酸培氟沙星原料药收载于EP9.0,USP及JP均未收载该品种。有关物质是评价药物安全性的一个重要指标,也是评价药品质量的重要指标[4]。本文参照EP9.0[5]建立了甲磺酸培氟沙星胶囊含量及有关物质的测定方法。
1 仪器与试药
1.1 仪器。Agilent1100高效液相色谱仪(紫外检测器和DAD检测器),色谱柱:SHISEIDO(资生堂)MG5μm C18 4.6 mml.d.×250 mm。
1.2 试药与药品。乙腈、N,N,N-三甲基1-十六烷基溴化铵、硼酸均为国产分析纯,硫二甘醇为Johnson Mathey Company提供。甲磺酸培氟沙星胶囊(A厂,规格:0.2 g;B厂,规格:0.2 g)各三批。
2 色谱条件
流动相:乙腈-N,N,N-三甲基1-十六烷基溴化铵(2.70g/L)与硼酸溶液(6.18 g/L)的混合溶液(用1 mol/L氢氧化钠溶液调节pH值至8.30)-硫二甘醇(28:72:0.2);流速:1.0 mL/L;柱温为30℃;检测波长273 nm;进样量:20 µL。
3 方法学考察
3.1 专属性试验
3.1.1 破坏试验:取样品分别进行热破坏(60℃下放置10天)、酸破坏(加入1 mol/L盐酸)、碱破坏(加入1 mol/L氢氧化钠溶液)、光照破坏(裸置于强光下48小时)、紫外光破坏(裸置于紫外光下48小时)以及氧化破坏(加入30%双氧水1 mL),采用“2”的色谱条件进行杂质分离度的考察。结果显示主峰与各杂质均能较好的分离,且辅料对测定无干扰。分别精密吸取上述供试品溶液20 ul注入液相色谱仪,记录色谱图,见图1。
3.1.2 峰纯度检测:将“3.1.1”的破坏试验样品溶液用DAD检测器进行纯度检测,结果主峰纯度均大于990,结果表明,本品在酸、碱、高温、光照、紫外及氧化条件下均有新的杂质产生,但均与主峰有良好的分离度,表明本方法具有较好的专属性,能满足本品有关物质检查的要求[6-8]。

图1 峰纯度检测
3.2 检测限及定量限。取培氟沙星对照品约25 mg,用流动相溶解并稀释制成每1 mL中含培氟沙星0.2 mg的溶液,再稀释500倍,采用“2”的色谱条件进样,测得培氟沙星检测限为0.01989 ng(信噪比=3),定量限为0.08517 ng(信噪比=10)。
3.3 溶液稳定性。取培氟沙星对照品约25 mg,用流动相溶解并稀释至每1 mL中含培氟沙星0.2 mg的溶液,采用“2”的色谱条件分别在1小时、2小时、4小时、8小时、10小时进样,结果表明,溶液在室温条件下10小时内杂质含量基本未变,提示本品溶液能够在10小时内保持稳定。
3.4 回收率考察。对两个生产厂家的样品进行样品加标回收率试验。取培氟沙星对照品约25 mg,用流动相溶解并稀释至每1 mL中含培氟沙星0.2 mg的溶液,作为对照品溶液;取样品适量,用流动相溶解并稀释至对照品溶液相同浓度,作为供试品溶液。另分别配制相当于含量测定浓度80%、100%和120%的加标供试品溶液各三份,采用“2”的色谱条件测定其含量,扣除本底,计算回收率,并计算相对标准差(RSD),结果见表1。
结果表明,2个生产厂家的样品平均回收率分别为100.40%、100.59%,RSD分别为0.79%和0.77%,该方法测定含量准确可靠。
4 结果检测
按拟订的方法进行有关物质和含量检测,杂质峰出峰时间均在主峰前。结果显示,三个生产厂家9批样品采用自身对照法,单个杂质在0.10%-0.31%,均小于0.5%,总杂质在0.25%-0.52%,均小于1.0%。9批样品含量为95.2%-98.4%。
5 讨论
在277 nm和273 nm波长处,甲磺酸培氟沙星峰的响应值比较接近,在258 nm波长处,峰的响应值较低。经紫外扫描,原料及各种处方的制剂主峰最大吸收均在273 nm处,辅料在273 nm波长处几乎无吸收;经破坏试验结果可知,除了酸破坏,其余强制破坏样品均未在主峰后检出杂质峰,而EP9.0的检测方法中,258 nm波长处主要检测主峰以后出峰的杂质[9-11]。为了便于含量和有关物质检测方法操作的简便和一致性,最终确定检测波长为273 nm。
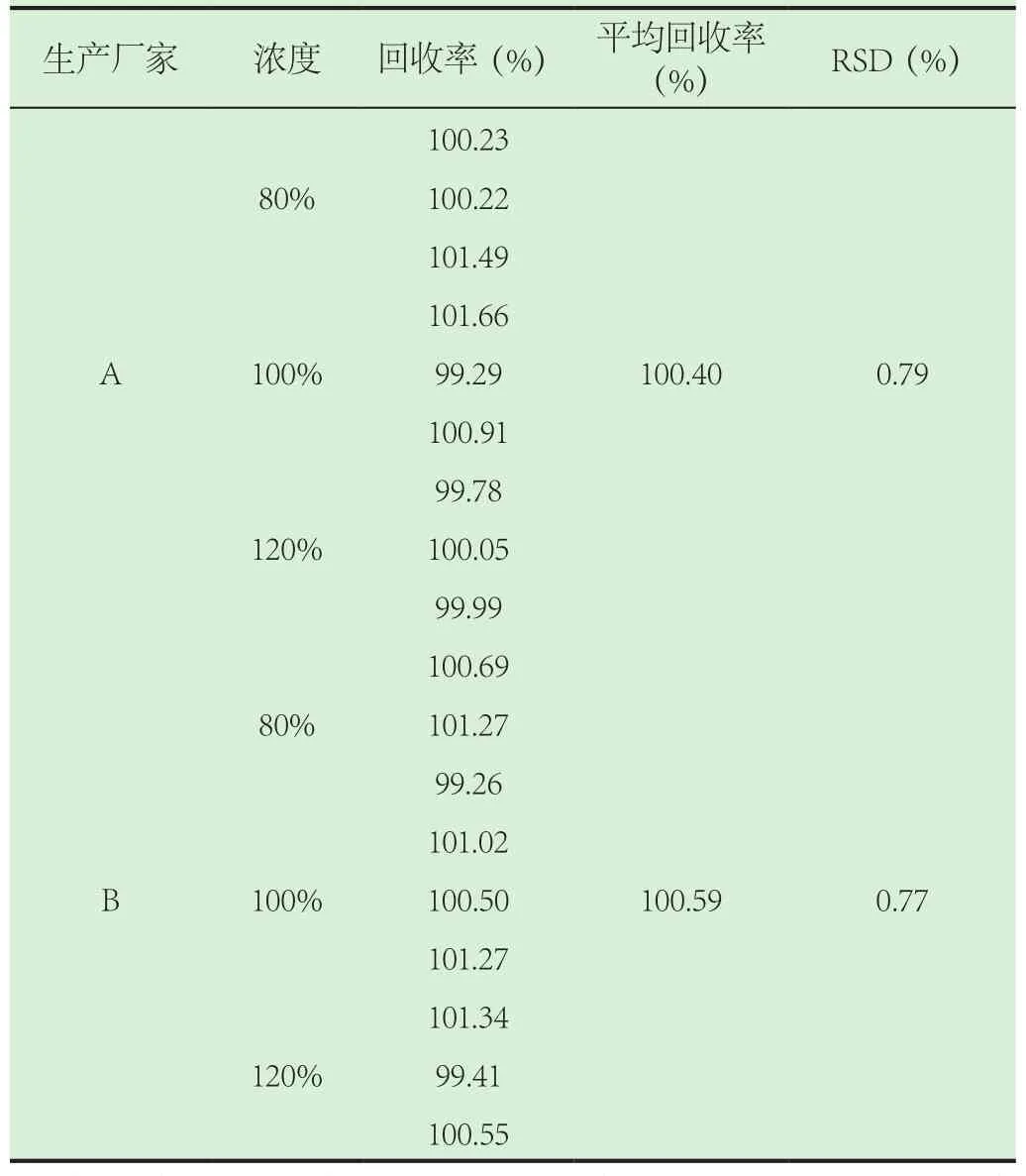
表1 回收率试验
甲磺酸培氟沙星对光较敏感,稳定性较差,在长时间强光和紫外光照射下,会产生较多杂质。因此在实验时可采取避光操作以降低环境因素对实验结果的影响。
