氧化微晶纤维素-壳聚糖接枝物的制备及表征*
2020-12-08陈淑花赵启成许利丽孙婷婷詹世平王景昌
陈淑花,于 驰,赵启成,许利丽,孙婷婷,詹世平,王景昌
(大连大学 环境与化学工程学院,辽宁 大连 116622)
0 引 言
壳聚糖和氧化微晶纤维素由于其自身的优异性能,比如生物降解性及相容性,对细胞无毒害等,引起了人们对生物高分子材料的极大兴趣,主要关于在医学领域的发展如:医用敷料、创伤修复、药物控释等[1-2]。
作为甲壳类动物骨架中最丰富的多糖之一,壳聚糖的来源十分广泛,通常存在于几种真菌和藻类的细胞壁中[3],壳聚糖具有无毒性,无致敏性,无致突变性;生物降解性较好,具有抗菌性;还具有资源丰富,廉价易得易贮存等特点[4-6]。由于纯壳聚糖膜具有较差的机械性能,这对其在医用敷料方面的应用有一定的限制。因此,已有实验将壳聚糖与天然或合成聚合物如纤维素等混合或交联等来改变其力学性能[7-10]。
通过对微晶纤维素本身化学结构的研究,发现纤维素分子结构中葡萄糖基环含有3个醇羟基,纤维素自身的分子与分子之间通过氢键结合,因此纤维素的物理性能十分稳定,具有结晶度较高以及机械性能较好等特点[11-12]。纤维素还具有良好的生物可降解性和生物相容性。但作为伤口敷料,它在生理液体中的溶解性受到一定的阻碍。因此,人们通过TEMPO三元复合氧化体系对微晶纤维素上的伯羟基改性,使其具备较大的比表面、高强度,低密度等特性,同时其氧化后的产物在溶液中具有良好的分散性,此外,它无毒,具有优良的生物相容性、血液相容性和生物降解性。同时,发现使用其氧化体系制备出的氧化微晶纤维素的羧基含量为16%~24%,止血性能最好[13]。
为了得到一种性能更好的生物材料,将CS与MCC(O)进行接枝,通过改变原料和EDC/NHS的质量比和反应温度,得到接枝率更高的产物,然后进行性能测定,以期得到一种新型生物医用材料。
1 实 验
1.1 实验原理
本实验的原理主要是将TEMPO自由基在NaClO的氧化作用下先生成一种亚硝翁离子,使亚硝鎓离子与纤维素结构单元上的伯羟基发生了亲核反应,从而选择性的使伯羟基变为了C6羧酸钠[14]。

图1 氧化微晶纤维素的制备原理Fig 1 Preparation principle of oxidation microcrystalline cellulose
使用EDC作为交联剂,NHS作为催化剂,将氧化微晶纤维素上的羧基与壳聚糖上的氨基形成酰胺键而结合。EDC/NHS组合在酰胺化反应过程中经常被使用,并且这两种试剂无毒,可以通过透析或冲洗轻松去除[15-16]。
1.2 实验试剂及仪器
微晶纤维素,分析纯,山东瑞泰化工有限公司;2,2,6,6四甲基哌啶-1-氧自由基,98%,上海阿拉丁生化科技股份有限公司;壳聚糖(5万),分析纯,南京奥多福尼生物科技有限公司;壳聚糖(20万、100万),DD≥95%,上海阿拉丁生化科技股份有限公司;1-乙基- ( 3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDC),分析纯,上海共价化学科技有限公司;N-羟基琥玻酰亚胺(NHS),分析纯,上海阿拉丁生化科技股份有限公司。
电子天平,BS200S,d=0.001 g,北京赛多利斯天平有限公司;数显恒温水浴锅,DF-101D,巩义市予华仪器有限责任公司;离心机,80-2,金坛市城西春兰实验仪器厂;循环水式真空泵,SHZ-D(Ⅲ),巩义市英峪仪器厂;真空干燥箱,DZF-6020,上海精宏实验设备有限公司;冷冻干燥箱,FD-1A-50,北京博医康实验仪器有限公司;红外光谱仪,Nicolet 560,美国尼高丽红外有限公司。
1.3 氧化微晶纤维素的制备
1.3.1 微晶纤维素的预处理
将5.0 g的微晶纤维素放入100 mL的NaOH溶液(质量分数为15%)中,在60 ℃下磁力搅拌2 h。然后将其冷却,抽滤,并用大量去离子水清洗,直至中性。将产物放在真空干燥箱中,在40 ℃下烘干备用。
1.3.2 微晶纤维素在TEMPO三元氧化体系中的改性
将2.0 g预处理的微晶纤维素放入Na2CO3/NaHCO3缓冲溶液(pH约为10.83)中,在40 ℃下高速搅拌使其溶解。随后按顺序加入40 mg TEMPO,0.6 g NaBr,继续搅拌,搅拌速度调为2 500 r/min,直至完全溶解。随后加入8 mL NaClO溶液(用HCL调节pH为10.0),然后每隔0.5 h加入一次NaClO溶液,控制速率,保持pH在10.0左右,反应5 h[17]。结束后将其倒入烧杯中,加入过量乙醇,得到白色析出物,用无水乙醇清洗数次,抽滤[18]。将产物在60 ℃下真空干燥,得到产物氧化微晶纤维素。
1.4 壳聚糖溶液的配制
移取50.00 mL 2%的HAC溶液,称取0.5 g壳聚糖放入其溶液中,在65 ℃下持续搅拌,使其完全溶解。
1.5 壳聚糖-氧化微晶纤维素接枝物的制备
在三颈烧瓶中分别加入0.5 g氧化微晶纤维素和50.00 mL去离子水,充N2除氧30 min,磁力搅拌使其充分溶解。随后加入一定质量比的EDC·HCL和NHS,在N2保护下搅拌活化30 min。然后向溶液中缓慢加入配好的壳聚糖溶液,使其充分混合,在一定温度下搅拌24 h,随后将溶液倒出,在溶液中缓慢滴加NaOH溶液,析出产物。然后,用去离子水洗涤至溶液为中性,离心干燥得到产物。
1.6 样品表征
1.6.1 产物的结构表征
红外光谱分析:使用傅里叶变换-红外光谱仪(FTIR)测定壳聚糖-氧化微晶纤维素接枝物的红外光谱图。采用溴化钾(KBr)压片法进行测试,波数范围为400~4 000 cm-1。
1.6.2 产物的接枝率检测
氧化微晶纤维素接枝物的接枝率为接枝支链占氧化微晶纤维素基质的质量百分数,按以下公式计算:
GR/%=(mG-mo)/mo×100%[19]
(1)
式中,GR为接枝率,mG为接枝产品的质量,mo为接枝前氧化微晶纤维素的质量。
1.6.3 产物的静态水接触角检测
将CS、MCC、CS-g-MCC(O)溶于2%的醋酸溶液中,加热搅拌使其充分溶解。然后将少量溶液均匀滴在载玻片上并干燥形成薄膜。取3 μL去离子水通过微量移液器滴加在待测膜的表面,使用仪器迅速拍下样品膜的接触角照片,并测量接触角的大小。
1.6.4 产物的热性能检测
产物的热性能通过差式扫描量热仪进行检测。首先分别准确称量10.0000 mg的CS、MCC、CS-g-MCC(O),小心放入坩埚内,设定初始温度为100 ℃,终止温度为350 ℃,升温速率为5 ℃/min。
1.6.5 产物的结晶性检测
使用X-Xay Diffraction对CS、MCC、CS-g-MCC(O)的结晶性能进行检测。使用Cu靶X射线管,扫描角度范围为5°~40°,扫描速度为1°/min。
2 结果与讨论
2.1 氧化微晶纤维素
2.1.1 氧化微晶纤维素取代度分析
在30 mL的丙酮中放入0.5 g氧化微晶纤维素,搅拌10 min,加入7.5 mL二甲基亚砜,以同样的转速搅拌1 h,充分溶解样品。以酚酞作为指示剂,用NaOH(0.1 mol/L)溶液标定,溶液变粉红色且不变色,记录消耗的NaOH溶液体积V0。持续搅拌,加入10 mL的NaOH溶液(1 mol/L),主要促进氧化纤维素醋酸酯水解,搅拌2 h,然后加入50 mL热水,使瓶壁周围冲洗干净,持续搅拌2~3 min。接着再加入2~3滴酚酞指示剂,用1 mol/LH2SO4溶液标定,溶液逐渐变为无色,再多滴加4滴H2SO4标准溶液,记录所消耗的H2SO4标准溶液的体积C。持续搅拌,保持溶液充分混合,5 min后,用0.1 mol/L的NaOH溶液标定,当溶液呈粉红色,并在1 min中内不褪色,将所消耗的NaOH体积记为A。同时按相同的步骤做空白试验。
取代度DS=[(D-C)Na+(A-B)Nb]×6.005/W[20]
(2)
式中:A为滴定样品所消耗的NaOH的体积,mL;B为滴定空白所消耗的NaOH的体积,mL;Nb为NaOH的浓度,mol/L;C为滴定样品所消耗的H2SO4的体积,mL;D为滴定空白所消耗的H2SO4的体积,mL;Na为H2SO4的浓度,mol/L;W为所用样品质量,g。
通过实验数据:W=0.200 g,D=6.68 mL,C=1.95 mL,A=4.52 mL,B=13.76 mL求出取代度DS=16.98%,因此本实验采用的氧化微晶纤维素平均取代度在16.98%左右。根据相关文献报道,当羧基的含量保持在16%~24%范围内的时候,氧化纤维素便具有十分优异的止血性能。因此,此实验所选用的氧化微晶纤维素具有十分优良的止血能力。
2.1.2 氧化微晶纤维素羧基含量分析
将0.3 g氧化微晶纤维素放入到5 mL的NaCl溶液(0.01 mol/L)与55mL的去离子水混合溶液中,用0.1 mol/L的HCL溶液调pH值,介于2.5~3.0之间,再用0.1 mol/L的NaOH溶液进行标定,用电导率仪器对滴定的曲线进行记录。
羧基含量(mmol/g)=C(V1-V2)/M×1 000[21]
(3)
式中:C为NaOH溶液浓度;V1为曲线平稳初期消耗的NaOH标液体积;V2为曲线平稳后期消耗的NaOH标液体积;M为氧化微晶纤维素的质量。

图3 电导率的滴定曲线图Fig 3 Conductivity titration curve
据文献报道,采用TEMPO氧化体系在pH为10时进行氧化反应,其氧化后的纤维素羧基含量可达到1.4~1.7 mmol/g[22]。结合图3和公式(3),可以求得氧化微晶纤维素羧基含量为1.56 mmol/g,和研究结果相符合。
2.2 壳聚糖-氧化微晶纤维素接枝物
2.2.1 红外光谱(FT-IR)分析
MCC(O)在1 740、1 638.05和1 414.33 cm-1出现-COOH的非对称伸缩和对称伸缩振动峰,在3 450 cm-1处出现O-H的伸缩振动吸收峰[22]。在上述CS光谱中,在3 432 cm-1处出现吸收峰,即为氢键缔合的O-H和N-H的伸缩振动吸收峰,1 638.18cm-1吸收峰归属为C=O的伸缩振动吸收峰[23]。而在MCC(O)-CS谱图中,在1 616.06、1 542cm-1处出现吸收峰,归属于酰胺键的酰胺I、II谱带,说明微晶纤维素氧化物和壳聚糖在羧基和氨基位置处通过形成酰胺键结合[24]。
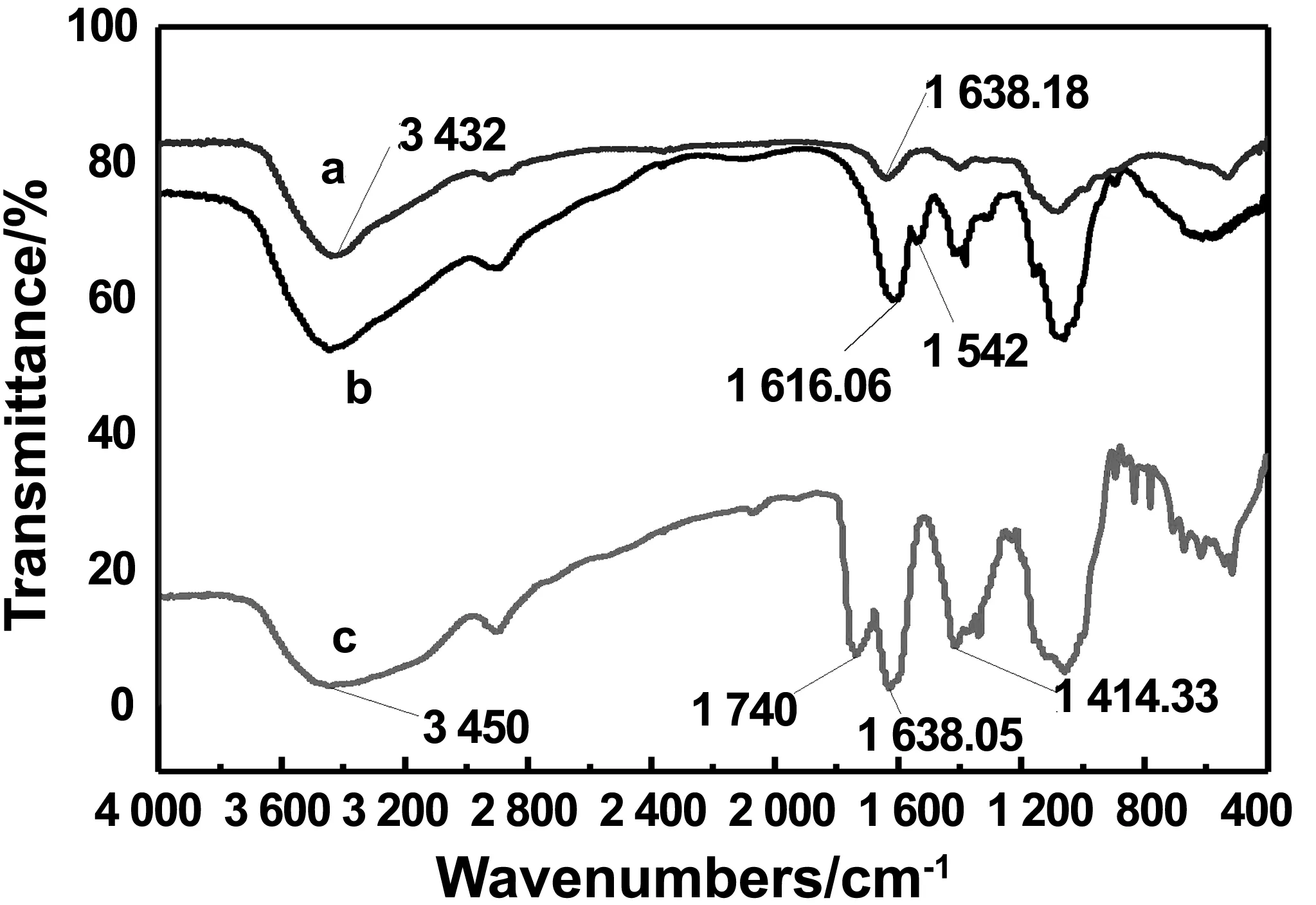
图4 接枝共聚物的红外谱图:(a)CS,(b)MCC(O)-CS,(c)MCC(O)Fig 4 FT-IR spectra of CS,MCC(O)-CS and MCC(O)
2.2.2 氧化微晶纤维素的接枝率
由表1可知,当壳聚糖分子量为5万,温度为60 ℃,氧化微晶纤维素和壳聚糖的含量为1∶2,EDC和NHS的含量比为3∶1时,其接枝率最大。

表1 氧化微晶纤维素的接枝率
2.2.3 X射线衍射(XRD)分析
壳聚糖的衍射曲线在12.596°处具有弱的衍射峰,在19.947°处具有尖而强的衍射峰,峰形尖锐,结晶度高[25];在12.105°、19.621°、21.619°处,氧化微晶纤维素显示不同强度的衍射峰。壳聚糖/氧化纤维素接枝物的衍射曲线在12.576°、19.866°、21.459°处显示不同强度的衍射峰,说明氧化纤维素的接入改变了壳聚糖原有的结晶性能,同时说明壳聚糖与氧化纤维素接枝聚合物的形成。图5中,壳聚糖/氧化微晶纤维素接枝聚合物的衍射峰变宽,强度明显减弱,说明其结晶性能减弱,无定形程度增大。这可能是因为壳聚糖的—NH3+与氧化微晶纤维素的-COO-之间发生静电作用,同时两者形成分子间氢键,限制了分子链的运动,在凝固再生过程中很难发生结晶[26]。

图5 接枝共聚物的XRD图谱:(a) CS, (b)MCC(O),(c)MCC(O)-CSFig 5 XRD spectra of CS,MCC(O) and MCC(O)-g-CS

图6 接枝共聚物DSC曲线图:(a)MCC(O)-CS, (b) MCC(O),(c) CSFig 6 DSC spectra of MCC(O)-CS,MCC(O) and CS
2.2.4 热稳定性测试
由图6可发现,壳聚糖、氧化微晶纤维素、壳聚糖/氧化微晶纤维素的最高分解温度分别为260、310、310 ℃;接枝物的热流量高于氧化纤维素的热流量,说明接枝物在分解的过程中需要吸收的能量越多,这表明接枝物的热稳定性能更强。这表明在氧化微晶纤维素/壳聚糖接枝物中的两种组分间的相互作用使得分解氧化微晶纤维素和壳聚糖分子中的糖苷键、C—H键、C—O键和C—C键更加困难,使其脱水、脱羧和脱羰难以进行,从而提高了热分解温度,热稳定性能得到改善。
2.2.5 静态接触角测试
接触角主要反映材料本身的亲水性,其大小表明膜材料是否具有亲水性。如果接触角越小,表示亲水性较好,制备的膜具有优异的亲水性,更好地吸收伤口渗出物,减少伤口表面细胞的粘连,伤口愈合加速。

图7 接枝共聚物的接触角液相图Fig 7 Contact angle liquid phase diagram of CS, MCC(O)-CS and MCC(O)
图7即为壳聚糖、氧化微晶纤维素/壳聚糖接枝物、氧化微晶纤维素的接触角图片,其都具有亲水性,通过将壳聚糖接枝到氧化微晶纤维素上,提高了其亲水性能,使性能得到了一定的改善,有利于伤口敷料的使用。
3 结 论
(1)为了优化CS与MCC作为单体的性质,通过EDC/NHS缩合体系制备CS-g-MCC(O)接枝共聚物,通过红外和XRD测试,证实了壳聚糖-氧化纤维素接枝聚合物的形成。
(2)由XRD测试结果可知,氧化纤维素的接入改变了壳聚糖原有的结晶性能,其结晶性能减弱,无定形程度增大。
(3)通过DSC测试,发现接枝物的热流量高于氧化纤维素的热流量,表明其接枝物的热稳定性能更强。
(4)通过接触角分析,发现接枝物的亲水性能得到提升,可以更好的吸收伤口渗出物,利于伤口愈合。
(5)通过以上测试结果可知,将生物相容性良好的壳聚糖和氧化微晶纤维素进行接枝,改善两者的不足,生成性能更为优异的生物医用材料,促进了生物医用材料的发展。
