基于生物信息学分析的口腔鳞状细胞癌微小RNA预后模型
2020-12-08赵格黎昌学郭超朱慧
赵格 黎昌学 郭超 朱慧
石河子大学医学院第一附属医院口腔科,石河子 832000
口腔癌是全球第九大恶性肿瘤,其中90%以上是口腔鳞状细胞癌(oral squamous cell carcinoma,OSCC),5年生存率仅为50%[1]。TNM分期是口腔癌的关键预后因素[2]。然而,由于高度异质性,TNM分期不能描述同一分期患者的个体风险。因此,需要新的生物标志物来区分高危患者,以帮助指导治疗。
微小RNA(microRNA,miRNA)是长度为18~25个核苷酸的非编码RNA,通过与其靶向mRNA的3’-非翻译区结合来调节基因表达,导致了mRNA降解或抑制mRNA翻译[3]。越来越多的研究[4-5]显示,miRNA在肿瘤细胞的生长、分化、增殖和凋亡等过程中发挥了重要的作用。部分miRNA已作为生物标志物开始应用到OSCC的诊断及预后判断[6-7]。
近年来,人们对癌症预后生物标志物进行了大量研究。与单一的生物标记物相比,多个基因的组合在预测个体预后方面显示了它们的优势[8-9]。本研究基于癌症基因组图谱(The Cancer Genome Atlas,TCGA)数据库,通过单因素和多因素Cox风险回归分析筛选和建立miRNA预后模型,以期对OSCC患者进行精准分组并为治疗提供依据。
1 材料和方法
1.1 数据下载
截止于2020年2月9日从TCGA数据库下载OSCC相关miRNA表达信息(398个肿瘤样本和32个正常样本)、mRNA表达信息(381个肿瘤样本和32个正常样本)和相关临床资料(401例)。从miRBase网站下载所有成熟miRNA序列,用Perl语言将其与差异表达miRNA合并。
1.2 差异表达分析
将下载的原始数据标准化处理后进行log2转换,使用R语言edgeR包比较肿瘤组与正常组miRNA和mRNA的表达差异,最终选取衡量错误发现率的指标(false discovery rate,FDR)<0.05,|log2FC|>1(FC为差异倍数,fold change)的差异基因与生存时间≥30 d的患者临床信息(共388例年龄为19~88岁患者,表1)相结合,建立标准化的miRNA和mRNA表达谱。
1.3 预后模型的构建及验证评估
通过R语言caret软件包将miRNA表达谱的样本随机分为train和test两组。对train组进行单因素Cox回归分析,筛选出P<0.01的miRNA。为了筛选并剔除引起多重共线性的miRNA,利用R语言survival包中“Coxph”和“direction=both”函数对单因素Cox回归得到的miRNA进行逐步多元Cox回归,最终建立生存相关预后模型。依据模型计算各组患者的风险评分,以风险评分中位值将患者分为高风险组和低风险组。Kaplan-Meier法绘制生存曲线并行Logrank检验(P<0.05)。使用survivalROC软件包计算患者5年生存率受试者工作特征曲线(receiver operating characteristic curve,ROC)下面积(area under curve,AUC)来评估预后模型的有效性和敏感性。
1.4 独立预后分析
通过单因素、多因素Cox回归分析,计算年龄、性别、病理分级、临床分期、肿瘤大小、区域淋巴结转移、风险评分与患者生存率之间的关系。判定所建立预后模型是否可以作为独立的预后因素。
1.5 靶基因预测及功能富集分析
从miRTarBase、targetScan和miRDB 3个数据库预测预后模型的靶基因,用Perl语言选取至少2个数据库中预测到的靶基因与TCGA数据库下载分析的差异mRNA取交集。通过R语言clusterProfifiler包和org.Hs.eg.db包对这些交叉基因进行基因本体论(gene ontology,GO)功能注释分析和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路富集分析(P<0.05,q<0.05)。
1.6 中枢基因筛选
通过STRING数据库绘制蛋白互作网络(protein protein interaction network,PPI)用于揭示靶基因之间的关系,置信度参数为0.700。使用Cytoscape3.6.1及其插件cytoHubba筛选出前10个中枢(Hub)基因。
1.7 统计分析
所有的统计均以R语言3.6.2版本以及其附属包为基础进行。
2 结果
2.1 6-miRNAs预后模型建立
依据上述方法及条件筛选出314个差异miRNA,其中上调185个,下调129个。6 190个差异mRNA,其中上调3 344个,下调2 846个。将具有生存时间的388名患者随机分为train组(n=196)和test组(n=192)。train组通过单因素Cox回归分析得到12个与患者总生存率相关的miRNA(P<0.01),通过逐步多因素Cox回归分析剔除引起多重共线性的miRNA后建立6-miRNAs预后模型。风险评分公式=(0.284 428×miR-499a-5p)-(0.560 344×miR-379-5p)+(0.326 790×miR-654-3p)+(0.364 695×miR-539-3p)+(0.229 542×miR-137-3p)-(0.216 523×miR-20b-3p)。
2.2 6-miRNAs预后模型评价
计算train组、test组和所有样品中患者的风险评分。均以train组风险评分中位值0.950为界,将患者分为高、低风险两组。风险生存曲线显示train组、test组和所有样品中高风险组的总体生存率明显低于低风险组(图1)。train组、test组及所有样品组中患者5年生存率ROC曲线的AUC值分别为0.757、0.673和0.724(图2);生存状态图显示3组中高风险评分患者的死亡率明显高于低风险评分患者死亡率(P<0.05)(图3)。
2.3 独立预后分析
单因素Cox回归分析显示,6-miRNAs预后模型与患者的总生存率明显相关(P<0.001)(图4左)。多因素Cox回归分析表明,在考虑其他临床因素时,6-miRNAs预后模型评分可作为影响OSCC患者总生存率的独立预后因素(P<0.001)。此外,区域淋巴结受累情况也是独立的预后因素(P<0.05)(图4右)。
2.4 6-miRNAs预测模型靶基因预测
3个数据库预测结果显示hsa-miR-20b-3p、hsamiR-137-3p、hsa-miR-379-5p、hsa-miR-499-5p、hsamiR-539-3p、hsa-miR-654-3p分别有431、1 115、701、307、733、426个重叠靶基因。为进一步提高生物信息学分析的可靠性,与TCGA下载分析得到的差异mRNA取交集,最终得到目标靶基因348个。
2.5 靶基因的GO和KEGG通路富集分析
对348个靶基因进行GO注释。与生物学过程(biological process,BP)、细胞组分(cellular component,CC)和分子功能(molecular function,MF)相关的前15个注释结果绘成图。富集结果中BP主要包括跨突触信号调节、化学突触传递调节和突触组织,CC主要包括突触膜、突触前膜和谷氨酸能突触,MF主要包括细胞外基质结构组成、DNA结合转录激活活性和特异性RNA聚合酶Ⅱ、生长因子活性。KEGG通路富集集中在致心律失常性右室心肌病(arrhythmogenic right ventricular cardiomyopathy,ARVC)和细胞黏附分子(cell adhesion molecule,CAM)上(P<0.05,q<0.05)。
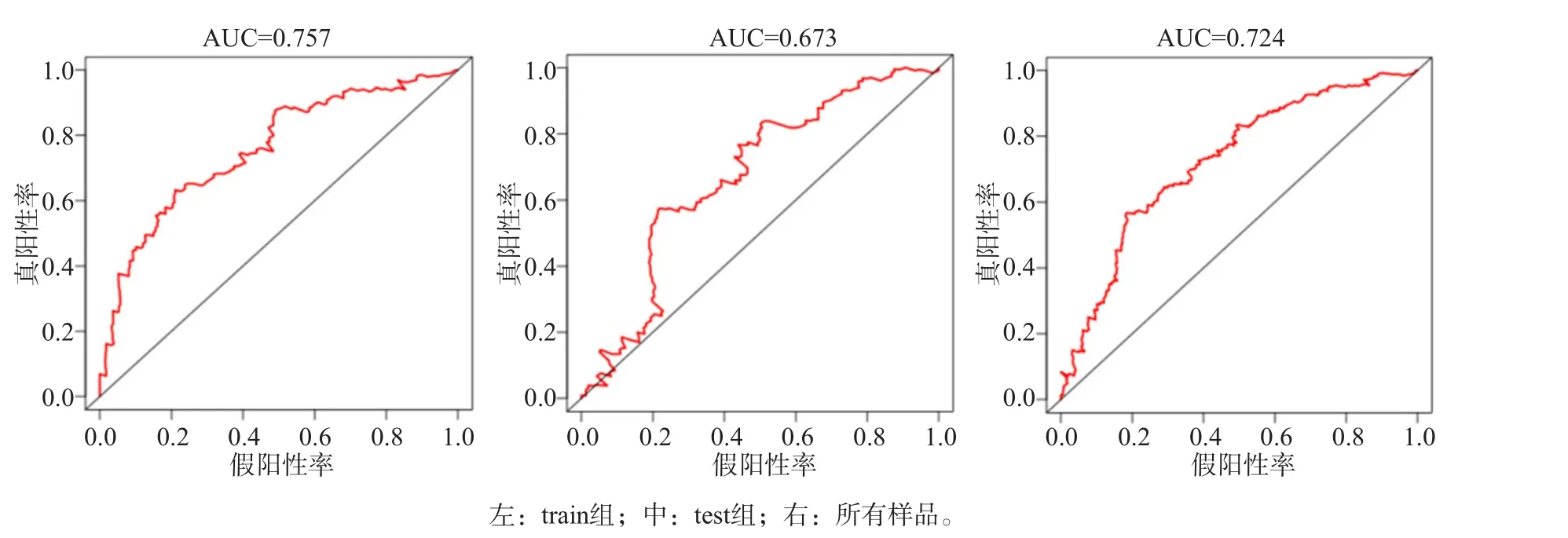
图2 患者5年生存率ROC曲线AUC值Fig 2 The AUC value of ROC curve of 5-year survival rate
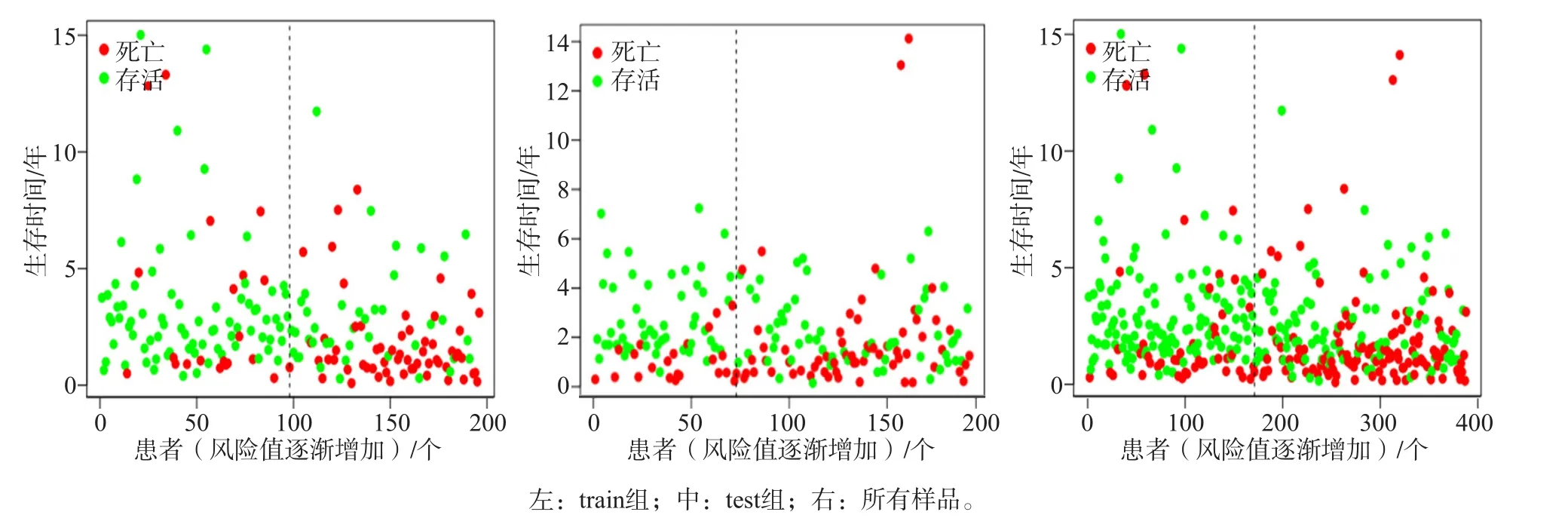
图3 高低风险组患者生存状态Fig 3 Survival status in high and low risk patients
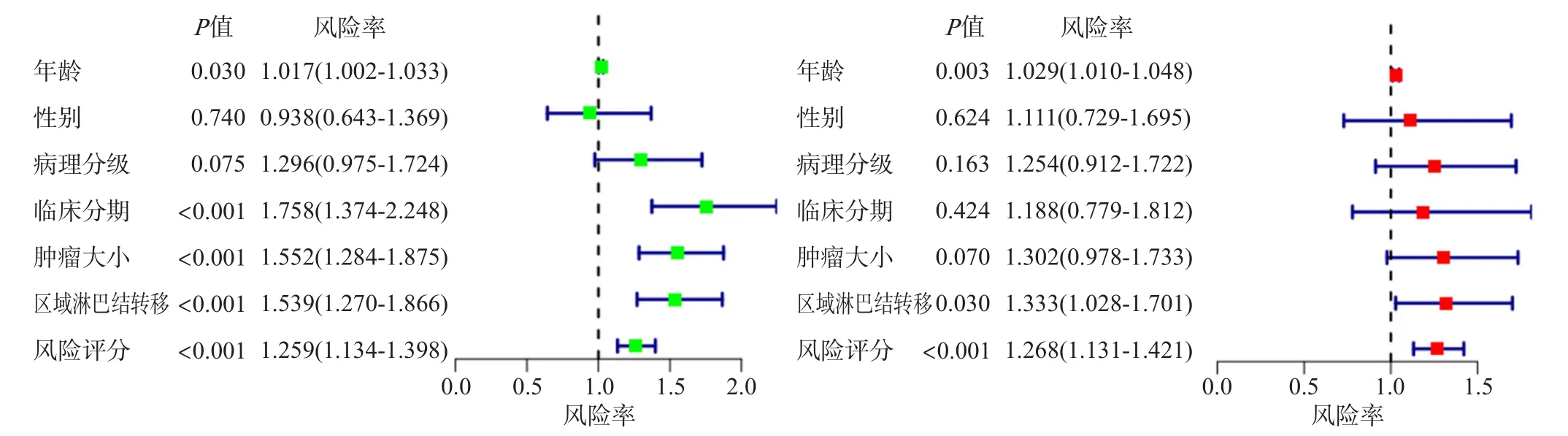
图4 单因素独立预后分析(左)和多因素独立预后分析(右)Fig 4 Univariate Cox regression (left) and multivariate Cox regression (right)
2.6 中枢基因的筛选
348个预测靶基因中,共有139个被过滤到靶基因PPI网络中,筛选得到10个中枢基因为CCNB1、EGF、KIF23、MCM10、ITGAV、MELK、PLK4、ADCY2、CENPF、TRIP13。其中EGF、ADCY2与患者生存相关(P<0.05)。
3 讨论
OSCC是世界范围内最常见的癌症类型之一。目前除了传统TNM分期外,缺乏能将高危患者进行区分的风险评估方法。近些年来,尽管努力改善影像学诊断技术和手术治疗方式,患者5年生存率仍未有明显改善[10-11]。因此,探索能够预测OSCC患者预后风险的预后模型具有重要意义。本研究从TCGA数据库下载并筛选OSCC差异miRNA,通过Cox单因素和多因素回归分析,构建了OSCC预后模型。发现由hsa-miR-379-5p、hsa-miR-499a-5p、hsa-miR-654-3p、hsa-miR-539-3p、hsa-miR-137-3p和hsa-miR-20b-3p组成的6-miRNAs预后模型可作为OSCC的独立预后因素。train组、test组和所有样品5年生存率ROC曲线的AUC值分别为0.757、0.673和0.724,一致显示该模型在预测OSCC患者生存风险方面具有较好预测能力,可作为可靠的预后模型。3组生存曲线显示高风险评分患者的死亡率均高于低风险评分患者,Log-rank检验差异有统计学意义。证明该模型可对OSCC患者进行精准的分组,或可提高患者治疗的针对性。
据报道,这6种miRNA参与了多种肿瘤的发生发展机制。miR-379-5p通过靶向调控MDM2的表达来抑制膀胱癌细胞的增殖、迁移和侵袭能力[12]。mir-499a-5p在高转移性肺癌细胞系及其外泌体中上调,促进细胞增殖、迁移和上皮间充质转化[13]。linc00460通过下调miR-654-3p在食管癌中发挥促癌作用,进而影响食管癌的预后[14]。miR-539-3p通过靶向sparcl1在上皮性卵巢癌中作为癌基因发挥作用[15]。胰腺癌中,circ-LDLRAD3下调通过miR-137-3p/ptn轴抑制胰腺癌细胞的增殖、迁移和侵袭[16]。miR-20b-3p参与了胶质母细胞瘤细胞中lnc-TALC介导的替莫唑胺耐药机制,为耐药性研究提供了新的思路[17]。这些研究成果一定程度上与本研究的模型相吻合。
为了进一步了解6-miRNAs预后模型在OSCC中的调控机制,本研究对预后模型的靶基因进行了功能富集分析。GO注释显示靶基因主要定位于突触膜,参与跨突触信号、化学突触传递的调节和突触组织组成;主要结合细胞外基质结构,具有生长因子活性、DNA结合转录激活活性和特异性RNA聚合酶Ⅱ等功能。KEGG通路富集集中在ARVC和CAM。研究[18]发现,CAM可将肿瘤细胞从原发灶中分离出来,减少细胞间黏附促进头颈部鳞状细胞癌的转移。这些富集结果都不同程度地显示了它们对肿瘤的作用,提示构建的miRNA预后模型可能参与肿瘤信号通路的调控。
为了寻找调控OSCC的miRNA预后模型关键节点,根据Cytoscape3.6.1及其插件cytoHubba筛选10个枢纽基因,分别为CCNB1、EGF、KIF23、MCM10、ITGAV、MELK、PLK4、ADCY2、CENPF、TRIP13。其中ADCY2、EGF与患者的生存相关;ADCY2参与膜受体的信号转导;EGF诱导上皮向间质转化,使头颈部癌细胞具有干细胞样表型[19]。因此,该预后模型可能通过调节ADCY2、EGF影响OSCC患者的生存及预后。
本研究的创新点在于以miRNA成熟体为研究对象且通过样本分组来验证模型可靠性。不足之处在于仅通对TCGA下载的的miRNA、mRNA表达谱进行大数据分析,而未进行实验验证。后期研究将收集病例进行实验验证。
综上所述,本研究得到了一个6-miRNAs独立预后预测模型,该模型可能有助于临床决策,确定适当的治疗计划,从而改善OSCC患者的预后和生存率。
利益冲突声明:作者声明本文无利益冲突。
