12q14微缺失综合征1例报告并文献复习
2020-12-03邹丽萍
陈 健 孟 岩 王 彬 张 培 杨 光 王 静 邹丽萍 刘 蕊
中国人民解放军总医院第一临床中心儿内科(北京 100853)
12q14微缺失综合征是一组以低出生体质量、智力低下、发育迟滞、身材矮小、学习障碍和脆性骨硬化症等为主要表现的疾病,主要由12号染色体长臂1区4带(12q14)微缺失所致[1]。国内至今未见相关报道。本文回顾分析1例与Silver-Russell综合征(SRS)有表型重叠且合并青春期早发育的12 q 14 微缺失综合征患儿的临床资料,结合相关文献进行复习,以提高对12q14微缺失综合征的认识。
1 临床资料
患儿女性,9岁3月龄,因身材矮小9年余、乳房发育伴快速生长1年余就诊。患儿G3P3,足月顺产,出生后即发现身材瘦小(具体数值不详),生后无追赶生长,生长发育明显落后于同龄儿。患儿自幼无喂养困难,喜食肉类,3个月抬头,5个月翻身,6个月独坐,1岁独走,1.5岁会说话,目前仍吐字不清,言语少,胆怯,能简单交流。现小学二年级,注意力不集中,学习成绩差。就诊前1年余患儿出现乳房发育,无月经初潮。近1年患儿身高增长20 cm。其母身高155 cm,15岁月经初潮;父身高160 cm;大姐 23岁,身高155 cm;二姐21岁,身高154 cm。家庭成员均体健,无智力落后者。体格检查:身高113 cm(-3.6 SD),体质量18.4 kg(-2.1 SD),头围45 cm(-4.2 SD)。幼稚面容,表情自然,全身皮肤干燥,有细小脱屑,双侧小腿伸面皮肤呈鱼鳞样改变。面部不对称,左侧较右侧偏小,小头,倒三角脸,小下颌,左侧上眼睑下垂,左眼内斜视,鼻梁宽鼻根部隆起,双耳不对称,左侧耳廓异常,呈杯状耳,双唇薄,腭弓高,牙齿未出齐,齿缝大。双手细长,小鱼际呈萎缩性改变,尺侧直平。双侧乳房对称性增大,Tanner分期Ⅲ期,乳晕色素加深。心前区无隆起,心浊音界正常,律齐,胸骨左缘2、3 肋间闻及连续机器样杂音,心尖区闻及Ⅱ/6级舒 张 期吹风样杂音,无心包摩擦音。脊柱侧弯,四肢对称、活动自如,站立、行走、跑跳均无异常。实验室检查:血常规、血生化、尿粪常规、甲状腺功能均无异常。血清类胰岛素生长因子-I 205 ng/mL,血清黄体生成素8.55 mIU/mL,血清卵泡刺激素11.06 IU/L。胰岛素、精氨酸生长激素激发试验中,生长激素峰值为2.14 μg/L。心电图、肝胆胰脾超声。双手及腕骨龄相当于11 岁儿童(图1 A)。头颅磁共振(MRI)示垂体前叶形态不规则,高度信号未见异常,后叶正常短T1信号存在,垂体柄未见增粗或偏移,鞍隔无抬高,鞍上池未见异常信号(图1B)。超声心动图示动脉导管未闭。双肾超声示左肾未见异 常,右肾位于脐水平,大小约6.9 cm × 4.5 cm × 2.8 cm,实质回声增厚、减低,肾内结构不清楚,集合系统比例 偏小;彩色多普勒血流成像(CDFI)检查可见两支动脉汇合入右肾动脉,集合系统内血流信号稀疏。胸腰椎平片示胸腰椎生理曲度存在,胸腰椎序列正常,见侧弯,未见滑脱。妇科超声示子宫前位,约2.0 cm × 0.9 cm × 1.8 cm,宫颈长约1.4 cm,肌壁回声均匀,内膜隐约可见,似呈线状,宫腔内未见明显异常回声;右卵巢大小约1.6 cm × 0.9 cm ×0.8 cm,内可见>0.4 cm的卵泡约2个;左卵巢大小约1.6 cm × 1.0 cm × 1.0 cm,未见>0.4 cm的卵泡。
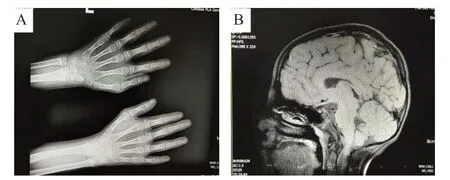
图1 患儿影像学表现
考虑到患儿重度身材矮小合并性早熟且伴多器官异常,遗传性疾病可能性大,需除外染色体病、微缺失/微重复综合征或单基因病可能,从经济学角度考虑行染色体微阵列分析(chromosomal microarray analysis CMA)。经医院医学伦理审核以及获得家长知情同意后,抽取患儿外周血3 mL,送检CMA。结果显示,患儿12号染色体q14.2q15区带缺失1个拷贝,长度约为5.8 Mbp。缺失段包含基因TMEM5、SRGAP1、C 12 orf 66、C 12 orf 56、XPOT、TBK 1、RASSF 3、MIR548C、GNS、WIF1、LEMD3、MSRB3、RPSAP52、HMGA 2、LLPH、TMBIM 4、IRAK 3、HELB、GRIP 1、CAND1……。分子细胞核型:arr[hg19]12q14.2q15(64,138,535-69,938,072) ×1。患儿确诊为12q14微缺失综合征。
2 讨论
通过PubMed、万方数据库、中国知网共检索到12篇文献,共报道12q14微缺失综合征患者26例,男13例,女14 例,其中有8 例来源于3 个家系。见表1。26例加本例患儿共27 例中多见的临床表型为身材矮小(23例)、颅面畸形(22例)、小于胎龄儿(21例)、智力低下/发育迟滞(21 例)、喂养困难(15 例)、语言发育延迟(12例)、学习困难(10例)、脆性骨硬化症(6例)、先天性心脏病(6 例),其他临床表型有相对大头畸形(5例)、脊柱侧弯(4例)、孤独症(4例)。
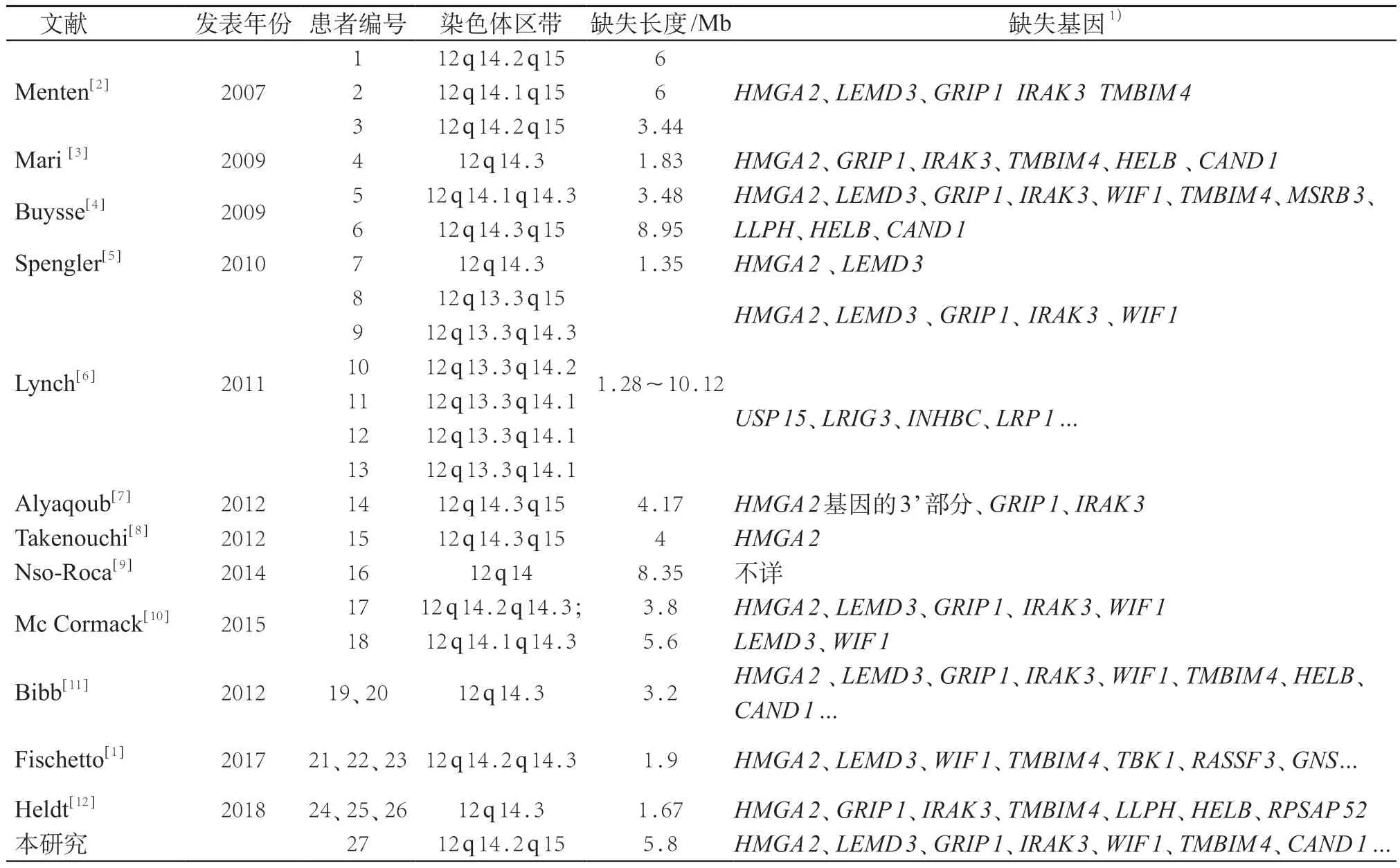
表1 12q14综合征患者染色体微缺失区带及缺失基因
对12q14微缺失综合征患者进行分析发现,其与SRS重叠的临床表型有出生体质量低于-2 SD、颅面畸形、矮小、喂养困难,不同于SRS的表型有智力低下/发育迟滞、语言发育延迟、学习困难等。初诊的27例患者中9例曾拟诊SRS(占33.3%),提示在伴有智力低下/发育迟滞、学习困难的SRS患者中,如果7号和11p15号染色体无遗传/表观遗传学改变,需注意本病的可能。12 q 14 微缺失综合征于2007 年首次描述[2],其临床表型中,小于胎龄儿、身材矮小、颅面畸形等与SRS 的表型重叠[5,12-13],不易区分。SRS 是一组临床和遗传异质性疾病,以宫内及生后生长迟缓、典型的颅面畸形、肢体不对称、喂养困难、先天性指侧弯为主要表现[14-15]。SRS主要由父源性11p15.5上印记基因区域(imprinting control region,ICR)1的低甲基化及7 号染色体母源性单亲二倍体(maternal uniparental disomy for chromosome 7,mUPD 7)所致[16-17]。此外12q14微缺失综合征还需与Dubowitz综合征相鉴别,后者具有面部畸形、低出生体质量、小头畸形、矮小及轻至中度发育迟缓等特征,55%的患者有潜在的遗传或基因组异常,如基因拷贝数变异,特别是在14 q 32和17q24处的缺失,或者单基因疾病包括新基因或双等位基因的致病性变异,如NSUN 2和LIG 4基因,但目前还没有一个单一的基因能够作为Dubowitz 综合征确诊指标,确诊多依赖临床特征[18]。12q14微缺失综合征发病机制为染色体微缺失,可依靠CMA确诊。
12 q 14 微缺失综合征是染色体微缺失所致疾病,在已报道患者中发现,其缺失区带多包含第12 号染色体长臂1 区4 带3 亚带,推测其临床表型相关基因多集中在3亚带。目前与临床表型可能相关的基因有HMGA2、LEMD3、IRAK3、GRIP1。
高迁移率的Hook2(HMGA2)基因是一种调节多种基因转录的结构因子,在胎儿组织中普遍高表达,在胚胎发育过程中起重要作用[19],在调节人类线性生长中也起着重要作用。动物实验证明,HMGA 2缺失可致侏儒表型即生长迟缓[20],对HMGA2基因缺失家族的相关研究也证明,缺失或单倍不足均可导致身材矮小[1,4-11]。在本研究总结的27例12q14微缺失综合征患者中,身材矮小的23 例均有HMGA 2缺失,仅4例无矮小者无HMGA 2的缺失。支持HMGA 2在身高生长中的重要作用,HMGA 2缺失与12 q 14 微缺失综合征的矮小、小于胎龄儿的临床表型相关[3,6-7,10]。
LEMD33基因参与骨形态发生蛋白信号转导,在脊椎动物胚胎发育中起到关键作用,在脆性骨硬化症家族中也发现了LEMD3变异[21-22]。在本研究总结的27例12q14微缺失综合征患者中,6例确诊脆性骨硬化症,年龄12~45 岁,均有LEMD 3基因缺失[1-2,11]。有8 例LEMD 3基因缺失者未发现骨骼病变[2,4-5,11],年龄1~16岁,其中6例年龄<10岁。脆性骨硬化症的发生是否与年龄相关目前尚不清楚,但建议动态监测LEMD3基因缺失患者的骨骼情况。
IRAK3编码与白细胞介素1受体相关的激酶,是先天免疫信号转导过程的负调节因子,对先天免疫的负性调节作用可能有助于防止过度炎症,但也可能使对肿瘤细胞的免疫监视功能下降,有助于肿瘤的形成和生长[23]。在动物实验中,IRAK3纯和缺失小鼠与体型减小和骨骼异常有关。有文献报道,IRAK 3疑似与脊柱侧弯表型相关[12]。在本研究总结的27 例患者中,4 例脊柱侧弯,其中3 例伴有IRAK3基因缺失,但在IRAK 3缺失的13 例患者中未出现脊柱侧弯表型,故IRAK 3缺失与脊柱侧弯表型是否相关有待进一步研 究。
谷氨酸受体相互作用蛋白1(GRIP1)在成人和胎儿大脑中高度表达,并参与谷氨酸突触传递,在早期神经元发育中起一定作用,被认为是发育迟滞和学习困难的候选基因[4,5]。本研究总结的27 例患者中,21例有智力低下/发育迟滞表型,且均有GRIP 1基因缺失。6 例智力正常患者中,5 例不伴GRIP 1基因缺失。因此推测GRIP 1基因缺失可能与发育迟滞和学习困难相关。
12q14微缺失综合征治疗主要是对症治疗。针对最常见的临床表型身材矮小,应监测身高,完善生长激素激发试验,对于生长激素缺乏患儿建议给予生长激素治疗,提高终身高。本研究总结的27例患者中,7例确诊生长激素缺乏,4例治疗有效,1例(1岁半)生长速度无变化,2例应用时间短未评价。本例确诊患儿还合并性早熟,2014年也有文献报告1例合并性早熟的12q14微缺失综合征[9],提示对于12q14微缺失综合征患儿仍需注意监测青春期发育情况。
综上,12 q 14 微缺失综合征最常见的临床表型是身材矮小,需监测患儿身高、生长激素水平及青春期体征,必要时给予生长激素治疗以提高终身高。对LEMD 3基因缺失的患儿需监测骨骼情况,以便早期发现脆性骨硬化症。因12q14微缺失综合征与SRS部分表型重叠,对没有典型特征的特发性SRS患者尤其是伴有智力障碍、发育迟缓者建议进行CMA。
