奥硝唑红外光谱的密度泛函理论研究
2020-11-30孙晓伟王文豪梁小蕊
孙晓伟 王文豪 梁小蕊


摘 要:运用密度泛函理论的方法在B3LYP/6-31G(d)水平上对奥硝唑红外振动光谱的特性进行研究。首先构建并优化分子几何构型,然后计算振动频率,并以此绘制红外振动光谱,最后对红外振动光谱的谱峰分布、谱峰的振动机理进行分析研究。研究结果表明:①0~1 800 cm-1区域内谱峰较多,其中1 000~1 800 cm-1区域内谱峰强度最强,400~1 000 cm-1區域内谱峰强度次之;②在1 700~3 000 cm-1区域内没有吸收,在 3 000~ 3 700 cm-1区域内有较弱吸收;③3 000~3 700 cm-1区域内谱峰分子振动形式以伸缩振动为主,0~1 800 cm-1区域内谱峰的振动形式多样。
关 键 词:奥硝唑;密度泛函;红外光谱;振动频率
中图分类号:O641.12+1 文献标识码: A 文章编号: 1671-0460(2020)09-1896-05
Abstract: The characteristics of ornidazole infrared vibration spectrum were studied by using density functional theory at the B3LYP / 6-31G (d) level. Its molecular geometry was built and optimized, the vibration frequency was calculated, and infrared vibration spectrum was drawn, spectrum peak distribution of infrared vibration spectrum and vibration mechanism of spectrum peak were analyzed and studied. The results showed that:①There were many spectral peaks in the region of 0 ~ 1 800 cm-1, among which the intensity of the spectral peaks was the strongest in the region of 1 000 ~ 1 800 cm-1, and the intensity of the spectral peaks in the region of 400 ~ 1 000 cm-1 was the second;②there was no absorption peak in the region of 1 700 ~ 3 000 cm-1, there was weak absorption peak in the region of 3 000 ~ 3 700 cm-1;③The molecular vibrational form of the spectral peaks in the region of 3 000 ~ 3 700 cm-1 was mainly stretching vibrations, and the vibrational forms of the spectral peaks in the region of 0 ~ 1 800 cm-1 were various.
Key words: Ornidazole; Density functional theory; Infrared spectra; Vibration frequency
奥硝唑为第三代硝基咪唑类抗菌药物,广泛应用于厌氧菌引起的感染病的治疗。与前两代硝基咪唑类药物相比,该药物具有良好的抗厌菌及抗滴虫作用,且具有疗效高、疗程短、体内分布广、副作用小等特点。目前,由于对奥硝唑分子结构和光谱特性缺乏深入研究,且硝基咪唑类化合物结构相似,典型药物检测方法难以对奥硝唑药物进行有效鉴别[1-8]。为此,本文采用广泛且有效的红外光谱分析法对奥硝唑分子几何构型和光谱谱峰分布等物质化学特性进行深入研究。
密度泛函理论是一种研究多电子体系电子结构的量子力学方法。近年来,密度泛函理论体系及其数值实现方法都有了很大的进展,使得该理论被广泛应用于化学问题的计算研究[9]。本文运用经典的Gaussian 09计算程序,在B3LYP/6-31G(d)水平上研究了奥硝唑分子的几何构型和红外光谱的谱峰分布特征,并对谱峰处分子的振动机理进行探讨。
1 计算方法
运用Chemoffice软件构建奥硝唑分子几何构型,然后采用Gaussian 09计算程序对其几何构型进行优化和频率计算,经频率分析保证计算所得频率中无虚频,进而确认计算所得构型为稳定构型。最后利用Gaussian view软件将频率计算所得数据转换为红外振动光谱进行分析。
2 结果与讨论
2.1 奥硝唑分子几何构型分析
用Chemoffice构建了奥硝唑分子的平面结构(见图1),用密度泛函B3LYP理论方法对奥硝唑分子进行了几何结构优化,图2给出了优化后的奥硝唑分子立体构型,一并标明了其原子编号及笛卡尔坐标系,表1给出了奥硝唑分子优化后的稳定键长、键角和二面角数据。由图2可见分子中的咪唑环在一个平面上,咪唑环与取代基—CH2—CHOH—CH2Cl之间有一定的扭曲,这一点可以通过表1中的二面角数据证实。从表1中可以看到二面角 ∠C1—N4—C15—C18、∠C3—N4—C15—C18和 ∠N4—C15—C18—C20分别为93.49°、84.36°和166.31°,除此之外其余的二面角均为接近180°或0°,充分说明咪唑环为一个平面,取代基—CH2—CHOH—CH2Cl在另一个平面上,二者之间的夹角为93.49°和84.36°,几乎呈互相垂直的状态;表1中的键角数据显示∠C8—C1—N6=126.3°、∠C8—C1—N4=126.9°、∠C2—C3 —N12=127.9°、∠C4—C3—N12=125.4°、∠C1—N4—C15 =125.0°、∠C3—N4—C15=125.8°,其余各键角均为110°左右,这说明奥硝唑分子中C1、C3和N4三个原子成键方式为sp2杂化,其余各原子为sp3杂化轨道成键;从表1的键长数据来看,优化后咪唑环中的C1—N6双键键长为0.135 0 nm,比咪唑单环中碳氮双键键长0.129 7 nm要长,N6—C2单键键长为0.139 3 nm,比一般咪唑中碳氮单键键长0.147 2 nm要短, C2—C3双键键长为0.138 9 nm,比一般咪唑中碳碳双键键长0.134 7 nm要长,C3—N4、N4—C1键长分别为0.142 4和0.136 4 nm,均比一般咪唑中碳氮单键键长要短,总的来说就是奥硝唑优化后,其咪唑环中单键键长缩短,双键键长增加,说明分子形成了共轭体系,使得其结构更加稳定。
2.2 奥硝唑红外振动光谱分析
利用物质分子对红外辐射的吸收,并由其振动或转动引起偶极矩的变化,产生分子振动能级和转动能级从基态到激发态的跃迁,得到分子振动-转动光谱,称为红外光谱。几乎所有的有机和无机化合物在红外光区均有吸收,除光学异构体、一些同系物以及某些分子量很大的高聚物外,凡是结构不同的两个化合物,一定不会有相同的红外光谱。红外光谱是定性鉴定化合物及其结构的重要方法之一,在化学、生命科学和环境科学等研究领域发挥着重要作用。
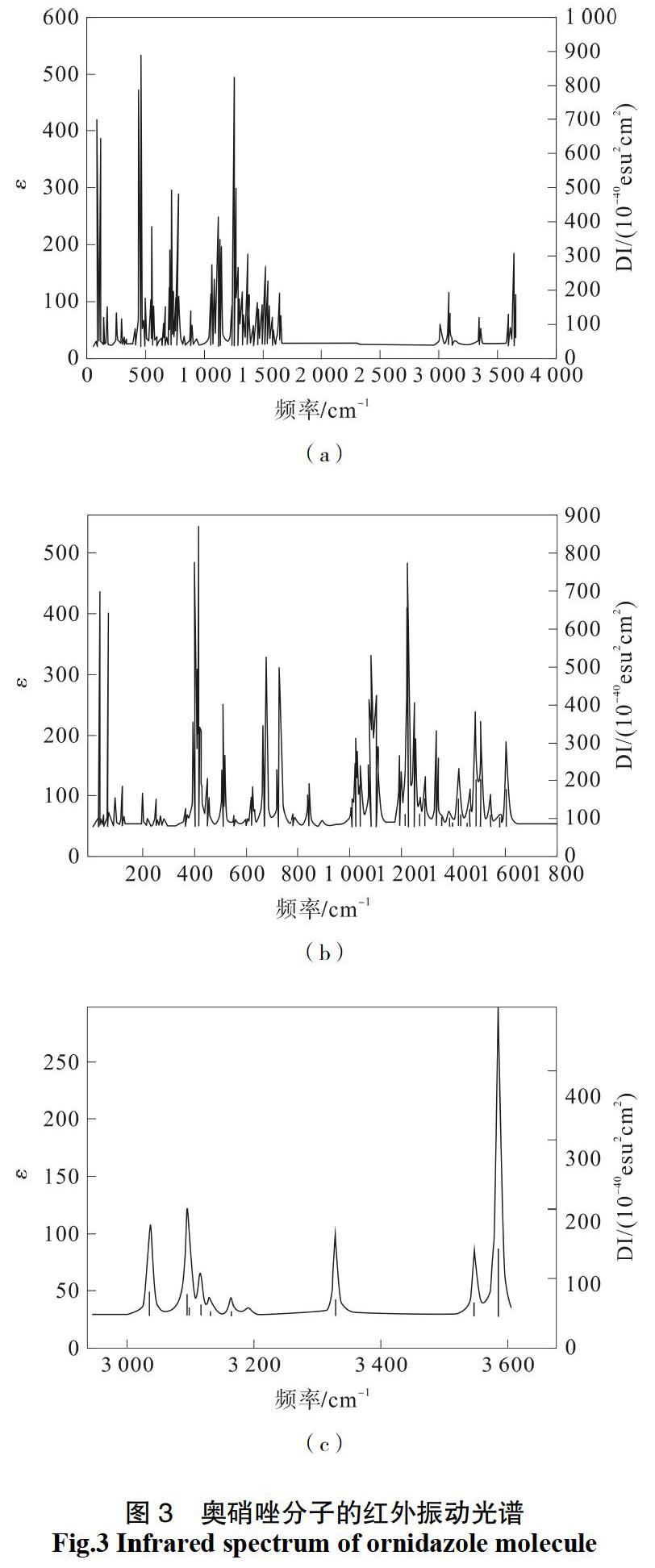
本文利用密度泛函理论方法计算了奥硝唑分子的振动频率,并利用Gaussian view软件绘制了红外振动光谱,如图3所示。图3中(a)是奥硝唑在0~3 800 cm-1范围内完整的红外谱图,为清楚起见,将其分为两部分,其中(b)是奥硝唑在0~1 800 cm-1处的谱图,(c)是3 000~3 700 cm-1处的谱图。
由图3可见0~1 800 cm-1区域内谱峰较多,对照振动频率数据分析,根据谱峰强弱的不同可将奥硝唑分子在这一区域的红外光谱分成三部分:0~400 cm-1、400~1 000 cm-1、1 000~1 800 cm-1。其中0~400 cm-1区域内谱峰强度都非常弱,不做讨论。400~1 000 cm-1区域内谱峰较强,这一区域的最强峰出现在422 cm-1处,它主要由24号氢原子的面外弯曲振动与16、17号氢原子的不对称弯曲振动共同作用引起的,如图4所示;次强峰出现在737 cm-1和682 cm-1处,它们分别由7号氢原子和5号氢原子的面外弯曲振动引起的,其分子振动示意图见图5、图6;在516 cm-1、675 cm-1处出现两个中等强度吸收峰,分别是由取代基—CH2—CHOH—CH2Cl上的17H、19H、21H、24H和21H、20C、22H的弯曲振动引起的,如图7所示。
1 000~1 800 cm-1区域内谱峰较400~1 000 cm-1区域的要稍强,最强吸收峰出现在1 241 cm-1处,是由21H、22H、19H、24H、16H、17H的面内摇摆振动和12N所在硝基的不对称伸缩振动引起的,见图8;1 023~1 209 cm-1区域的多个较强峰主要包括8号碳原子所在的甲基、20号碳所在的亚甲基和15号碳的亚甲基的不对称弯曲振动引起的; 1 506 cm-1处的较强峰是由21H、22H,16H、17H和10H、11H的对称弯曲振动引起的,见图9; 1 626 cm-1处还有一个中等强度的吸收峰,是由 2C—3C伸缩振动和5H、7H的面内弯曲振动引起的。
图3显示奥硝唑分子的红外光谱在1 700~3 000 cm-1区域内没有吸收,在3 000~3 700 cm-1区域内有较弱吸收,这一区域的分子振动模式主要以伸缩振动为主。最强吸收峰出现在3 681 cm-1处,是由分子中唯一的N—H伸缩振动引起的,见图11;几个中等强度的吸收峰位于3 042 cm-1、3 109 cm-1和 3 382 cm-1处,其中3 042 cm-1处的振动形式为8C所在甲基的对称伸缩振动,3 109 cm-1处的振动形式为16H、17H的对称伸缩振动和19H的伸缩振动,3 382 cm-1处的振动形式为5H的伸缩振动,如图12、图13、图14所示。
3 结 论
本文运用密度泛函理论得到了奥硝唑分子的几何构型及红外振动光谱,并在此基础上详细分析了奥硝唑分子的结构特点、各区域谱峰的分布情况及谱峰处分子振动机理。
通过对红外光谱的分析发现:0~1 800 cm-1区域内谱峰较多,其中1 000~1 800 cm-1区域内谱峰强度最强,400~1 000 cm-1区域内谱峰强度次之;在1 700~3 000 cm-1区域内没有吸收,在3 000~3 700 cm-1区域内有较弱吸收;3 000~3 700 cm-1区域内谱峰分子振动形式以伸缩振动为主,0~ 1 800 cm-1区域内谱峰的振动形式多样。本文研究可为奥硝唑药物的快速检测、硝基咪唑类化合物鉴别提供理论依据。
参考文献:
[1]田怀平,王美纳. 奥硝唑的药理作用及临床应用[J].中国医院用药评价与分析,2002,2(5):303-304.
[2]OBERLANDER G, YEUNG C H, COOPER T G. Influence of oral administration of ornidazole on capacitation and the activity of some glycolytic enzymes of rat spermatozoa[J]. Journal of Reproduction & Fertility,1996,2(2):231-239.
[3] 金春香.奥硝唑与替硝唑、甲硝唑治疗滴虫性阴道炎的疗效比较[J]. 中国社区医师(综合版),2006,8(13):45.
[4]张振伟,左剑,张存林. 甲硝唑、替硝唑和奥硝唑药品的远红外与太赫兹吸收光谱研究[J]. 光谱学与光谱分析,2012,32(4):906-909.
[5]KANTARJIAN H M, GILES F J, TSIMBERIDOU A M,et al. Pilot study of recombinant human soluble tumor necrosis factor (TNF) receptor (p75) fusion protein (TNFR:Fc; Enbrel) in patients with refractory multiple myeloma: increase in plasma TNF alpha levels during treatment[J]. Leukemia Research:A Forum for Studies on Leukemia and Normal Hemopoiesis,2003,27(5):375-380.
[6]杨玉平,陈笑,姚飞,等. 甲硝唑、替硝唑和奥硝唑的拉曼光谱研究[J].中央民族大学学报(自然科学版),2010,19(4):26-30.
[7]凌智鋼,唐延林,李涛,等. 甲硝唑、替硝唑和奥硝唑的结构与光谱[J].原子与分子物理学报,2013,30(4):553-558.
[8]陈蒂芳,易爱纯,张顺芝. UV法测定奥硝唑分散片的含量[J]. 中国药事,2004,18(1):44-45.
[9]余红静,刘照军,尹延峰,等. 甲醇锂和乙醇锂拉曼光谱的密度泛函理论研究[J]. 光谱学与光谱分析,2009,29(11):2975-2979.
