小麦主栽品种济麦22与良星99的基因组序列多态性比较分析
2020-11-27杨正钊王梓豪胡兆荣辛明明姚颖垠彭惠茹尤明山宿振起郭伟龙
杨正钊 王梓豪 胡兆荣 辛明明 姚颖垠 彭惠茹 尤明山 宿振起 郭伟龙
小麦主栽品种济麦22与良星99的基因组序列多态性比较分析
杨正钊 王梓豪 胡兆荣 辛明明 姚颖垠 彭惠茹 尤明山 宿振起*郭伟龙*
中国农业大学农学院/ 农业生物技术国家重点实验室/ 杂种优势研究与利用教育部重点实验室, 北京 100193
济麦22和良星99是我国黄淮冬麦区和北部冬麦区大面积推广的高产小麦品种, 也是目前小麦杂交育种的重要亲本。虽然济麦22和良星99的来源和系谱不同, 但在重要农艺、产量等性状上存在较高的相似性。为了从全基因组水平研究其遗传组成的异同, 本研究采用Illumina HiSeq2500测序平台对上述两个品种进行了全基因组测序(平均测序深度为5.8×), 并系统地比较了两个品种拷贝数变异(CNV)、单核苷酸多态性(SNP)和插入/缺失(InDel)的序列差异。与中国春参考基因组序列相比, 两个品种除了具有总长466 Mb的共有CNV变异区间外, 济麦22和良星99的特有CNV变异区间的总长分别为91 Mb和45 Mb, 这些特有CNV区间主要集中在2B和4B染色体上; 济麦22和良星99间存在1,547,371个SNP差异位点和137,817个InDel差异位点。以差异SNP分布规律为依据, 在全基因组水平鉴定出济麦22和良星99间存在14.2%的差异多态性热点区间, 这些区间集中分布在1D、2B和4B染色体上。通过对5个控制小麦株高和穗长基因的序列分析, 发现有2个位于多态性热点区间的基因在品种间存在移码突变。本研究为利用重测序数据在基因组水平上比较小麦品种间遗传差异提供了重要参考, 同时揭示了济麦22和良星99在全基因组的遗传相似区间和差异区间, 为今后小麦育种改良中更好利用济麦22与良星99提供了重要遗传信息。
小麦; 济麦22; 良星99; 全基因组重测序; SNP; CNV
小麦是重要的口粮作物。我国是世界上最大的小麦生产和消费国, 提高单产是保障我国小麦生产可持续发展和粮食安全的重要途径。新中国成立以来, 我国小麦主产区经历了5~6次的品种更换, 小麦品种的产量、抗性、品质等重要性状得到了大幅提高。小麦品种的遗传改良对我国小麦单产和总产的提高发挥了重要作用[1]。不同小麦生态区中对照品种既是各时代育种水平的代表, 通常又是下一代品种改良的骨干亲本[2]。例如, 黄淮冬麦区的对照品种周麦18[3]、鲁麦14[4]、石4185[5]等在小麦的遗传改良中做出重要贡献。
常规育种是我国小麦遗传改良的主要手段, 但在亲本选配、后代选择等关键育种过程中依赖于有限的表型性状和育种者的经验。育种过程中存在不确定因素和盲目性, 影响了小麦遗传改良的效率。分子标记的开发和应用为小麦性状的基因定位、标记辅助选择、亲本遗传多样性分析等方面提供了重要支撑, 可有效提升抗性、品质等性状遗传的改良效率。继水稻、玉米、大麦等主要作物基因组测序的完成, 六倍体普通小麦中国春的高质量基因组参考基因组序列(IWGSCv1)的测序完成, 标志着小麦遗传研究已进入“后基因组学时代”[6]。与基于表型和有限分子标记辅助的选择相比, 利用基因组测序并从全基因组水平解析品种间的遗传差异为小麦的遗传研究和品种的改良提供高维度的组学数据支持[7]。
济麦22是山东省农业科学院作物研究所选育[8]。良星99是山东省德州市良星种子研究所选育[9]。济麦22和良星99分别于2006年通过黄淮冬麦区北片审定, 此后相继通过北部冬麦区和黄淮南片等麦区审定, 累计推广面积超过6000万公顷。由于综合性状突出, 济麦22 (2015至今)和良星99 (2010—2015)先后作为黄淮冬麦区北片区域试验的对照品种, 也是我国当前小麦品种改良的重要亲本资源。育种家在长期育种和生产实践中发现, 虽然济麦22和良星99来源及系谱不同, 但两品种间除株高外, 田间总体形态相近; 主要农艺、产量和抗病反应等性状表现一致, 两者的杂交后代亦无明显分离, 普遍认为两个品种遗传组成相似性较高, 亲缘关系较近。近期研究发现麦22和良星99的2个衍生材料的白粉病抗性相近, 通过分子标记定位发现两者具有相同基因[10]。但对其遗传相似性的认识仍以有限的表型性状描述和经验为主, 尚未从遗传水平上对两者进行系统解析。
本研究通过对济麦22和良星99的重测序数据分析, 在全基因组水平系统解析了两者的遗传组成差异, 以期对今后小麦育种的亲本利用、重要性状基因区间追溯、新品种系谱分析、基因组水平上研究重要骨干种质的遗传组成等方面提供重要参考。
1 材料与方法
1.1 小麦材料及田间表型考察
供试材料为小麦品种济麦22 (系谱: 935024× 935106)与良星99 [系谱: (济91102×鲁麦14)×PH85-16]。2018年10月1日将上述两个品种播种于中国农业大学上庄实验站(北京, 40°14′N, 116°19′E), 按照完全随机试验设计, 设3次重复, 2行区, 行长1.5 m, 行距25.0 cm, 株距3.0 cm。收获前每小区取中部10株调查株高、穗长、小穗数、穗粒数、旗叶长和宽等农艺性状; 收获后利用考种仪(良田高拍仪S500A3B)考察千粒重、粒长、粒宽等籽粒性状。品种间性状差异检验用测验进行统计分析, 数据分析通过R语言计算完成。
1.2 全基因组重测序与多态性位点鉴定
用CTAB的方法分别从济麦22和良星99幼根提取DNA。用Illumina HiSeq2500测序平台采用双端测序的方法进行全基因组测序, 建库和测序由北京诺禾致源科技股份有限公司完成。对重测序原始数据用Trimmomatic软件(v0.36)[11]进行过滤后再使用BWA软件(v0.7.15)[12]的BWA-MEM工具将过滤后的数据在小麦参考基因组(IWGSCv1)[13]上进行比对, 选择保留存在“唯一最优匹配(Unique Best Hit)”的读段对; 最后利用与samtools (v1.4)[14]工具进行过滤。
利用GATK软件(v3.8)[15]中的HaplotypeCaller、GenotypeGVCF、SelectVariants和VariantFiltration等功能计算并过滤获得材料基因组上的高质量SNP和InDel位点。其中, SNP位点过滤参数为“QD < 2.0, FS > 60.0, MQRankSum < –12.5, ReadPosRankSum < –8.0, SOR > 3.0, MQ < 40.0, DP > 30||DP < 3”; InDel过滤参数为“QD < 2.0, FS > 200.0, ReadPosRankSum < –20.0, DP > 30||DP < 3”。对过滤后的SNP与InDel位点通过SNPEff软件(v4.3t)[16]进行注释。使用的中国春基因注释版本为IWGSC RefSeq v1.1 (https:// urgi.versailles.inra.fr/download/iwgsc/IWGSC_RefSeq_Annotations/v1.1/)。
1.3 拷贝数变异区间鉴定及比较分析
拷贝数变异(copy-number variation, CNV)是小麦基因组研究的主要多态性类型之一。本研究为鉴定供试材料相对于中国春参照基因组的CNV变异区间, 在分析中以1 Mb为单位将全基因组划分为小窗, 利用bedtools (v2.26.0)[17]计算两个品种的重测序比对读段在每个窗口内的“平均覆盖深度”(Depbin);并结合该材料的全基因组“平均读段覆盖深度”(Depave)进行归一化, 得到每个小窗的“平均相对覆盖深度”: Depbin/Depave。由于在CNV分析的结果中, 基因组序列丢失相比于序列插入更容易被检测, 本研究计算得到的CNV变异区间仅考虑相对于参照基因组低覆盖度(“丢失”)情况。下文中的CNV均指“丢失(低覆盖度)变异”。“相对平均覆盖深度”低于0.5的小窗被视为“CNV变异区间”(附图1)。CNV变异区间分析均由每个材料分别和“中国春”参照基因组的序列比较计算获得。其中, 在两个品种中同时存在CNV变异的区间被称为“共有CNV变异区间”。
1.4 品种间全基因组高频率SNP差异区段的鉴定
为鉴定两个品种的基因组序列差异区间, 本研究以1 Mb为单位对全基因组进行划分窗口, 对每个窗口中的单核苷酸多态性(single nucleotide polymorphism, SNP)的频率进行分析, 在每个窗口中统计两品种间存在差异的纯合SNP的密度分布(附图2)。这部分研究中仅考虑拷贝数正常的区间, CNV变异区间未被统计。考虑该密度分布具有明显的高斯混合分布的特点, 我们利用EM算法拟合出两个正态分布的模型, 并据此选择阈值=1.5 (差异频率约为3.1×10–5)作为2个分布的分隔边界。其中, 高密度SNP分布区间被认为两个材料的多态性热点区间;低密度SNP分布区间被认为两个材料的相似区间。
1.5 差异CNV区间和基因序列差异差异位点的验证
对济麦22与良星99基因组内的特有与共有CNV区间分别进行验证, 选取区间内2000 bp左右长度的序列设计染色体区间特异的引物对CNV区间进行验证。扩增引物由在线软件Primer 3.0设计, 其中引物长度范围为18~24 bp, GC含量范围在40%~60%之间, 退火温度范围为54~60℃, 扩增产物大小1~2 kb (表1)。以济麦22、良星99与中国春的DNA为模板进行PCR扩增, 通过有无目标扩增产物判断CNV是否存在。对与小麦株高相关的候选基因、的序列差异SNP位点, 设计出基因组特异引物(表1)进行PCR扩增并测序。PCR反应体系为20 μL, 包括10 μL的2X M5 HiPer plusHiFi PCR mix, 正、反向引物(10 μmol L–1)各1 μL, 150 ng μL–1模板DNA 2 μL, 用ddH2O补至20 μL。PCR扩增程序为95℃ 3 min; 95℃30 s, 56~57.4℃ 30~60 s (依引物退火温度以及目标序列而定), 72℃ 2 min, 35个循环; 72℃5 min。

表1 本研究使用的引物编号和序列
2 结果与分析
2.1 济麦22与良星99的农艺性状比较
田间观察发现2个品种株形相近, 两者除株高、穗长存在显著差异外(< 0.01) (图1), 旗叶大小、小穗数、千粒重、粒长、粒宽等主要农艺和产量相关性状差异均不显著(表2)。这表明济麦22和良星99在主要农艺和产量性状上存在较高的相似性。
2.2 济麦22和良星99全基因组多态性位点检测
利用Illumina HiSeq2500分别对济麦22和良星99进行了全基因组测序, 分别获得了88 Gb和128 Gb的重测序数据。通过和中国春参考基因组序列(IWGSCv1)进行测序数据的比对并去除重复序列, 选择“唯一最优匹配”的读段进行后续分析, 最终两品种的平均覆盖深度均为5.8×。经过SNP calling分析, 济麦22和良星99中分别鉴定出15,326,314个与15,060,004个和中国春参照基因组不同的高质量纯合SNP位点, 约占全基因组的0.109%和0.107% (表3)。其中, A、B基因组中的SNP的频率(0.130%~ 0.155%)明显高于D基因组(0.0224%~0.0225%)。两品种间基因型不一致的纯合SNP位点仅占总SNP位点个数的9.8%, 即超过90%的SNP是两个品种间共有的。同时, 在济麦22和良星99中分别鉴定出1,143,512与1,118,564个的InDel位点(表4), 分别占全基因组的0.0813%和0.0795%。与D基因组, A、B基因组中鉴定出的InDel频率更高。济麦22和良星99间差异的InDel多态性位点占鉴定出的InDel位点总数的11.8%。
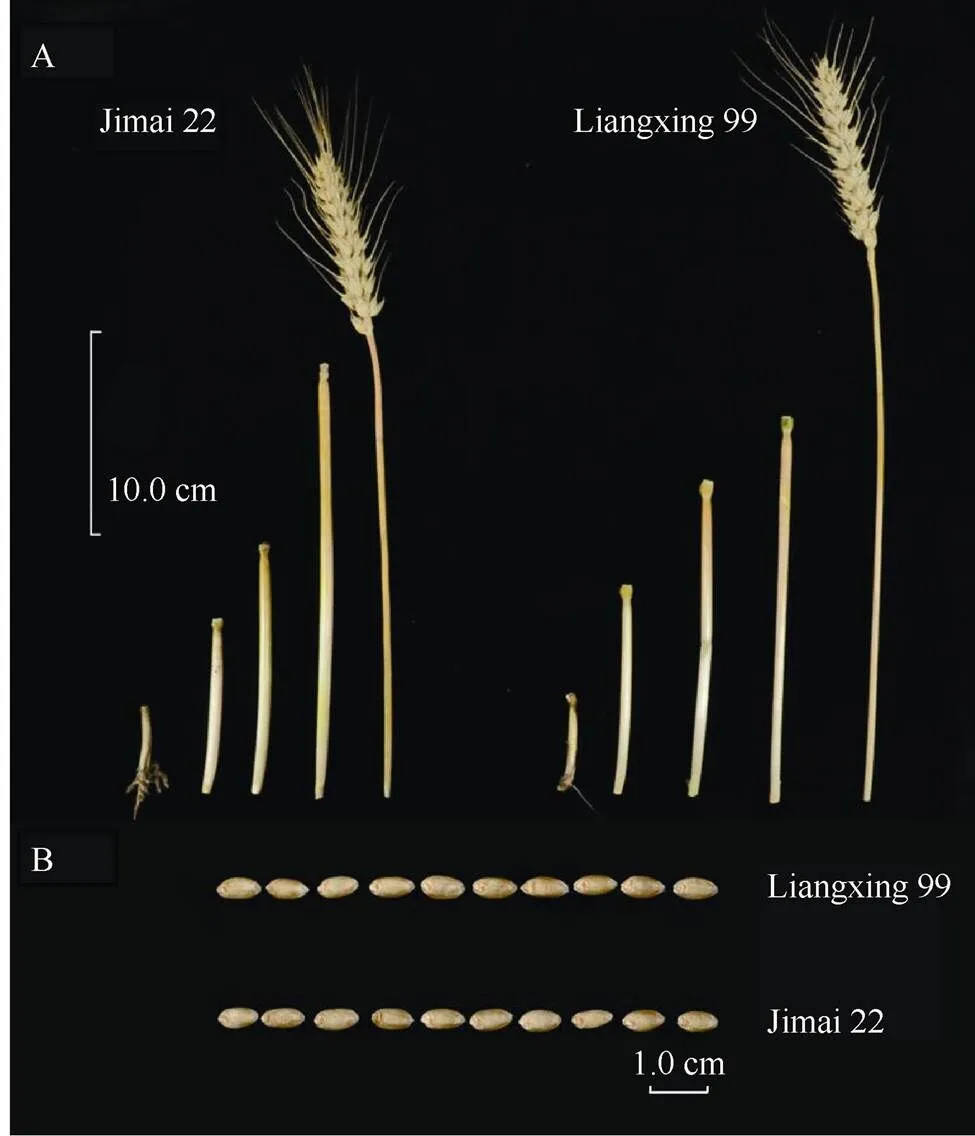
图1 济麦22与良星99的表型比较
A: 济麦22 (左)与良星99 (右)的株高及节间长度比较; B: 济麦22 (上)与良星99 (下)的粒型比较。
A: plant height and internode length comparison between Jimai 22 (left) and Liangxing 99 (right); B: grain morphological comparison between Jimai 22 (top) and Liangxing 99 (bottom).
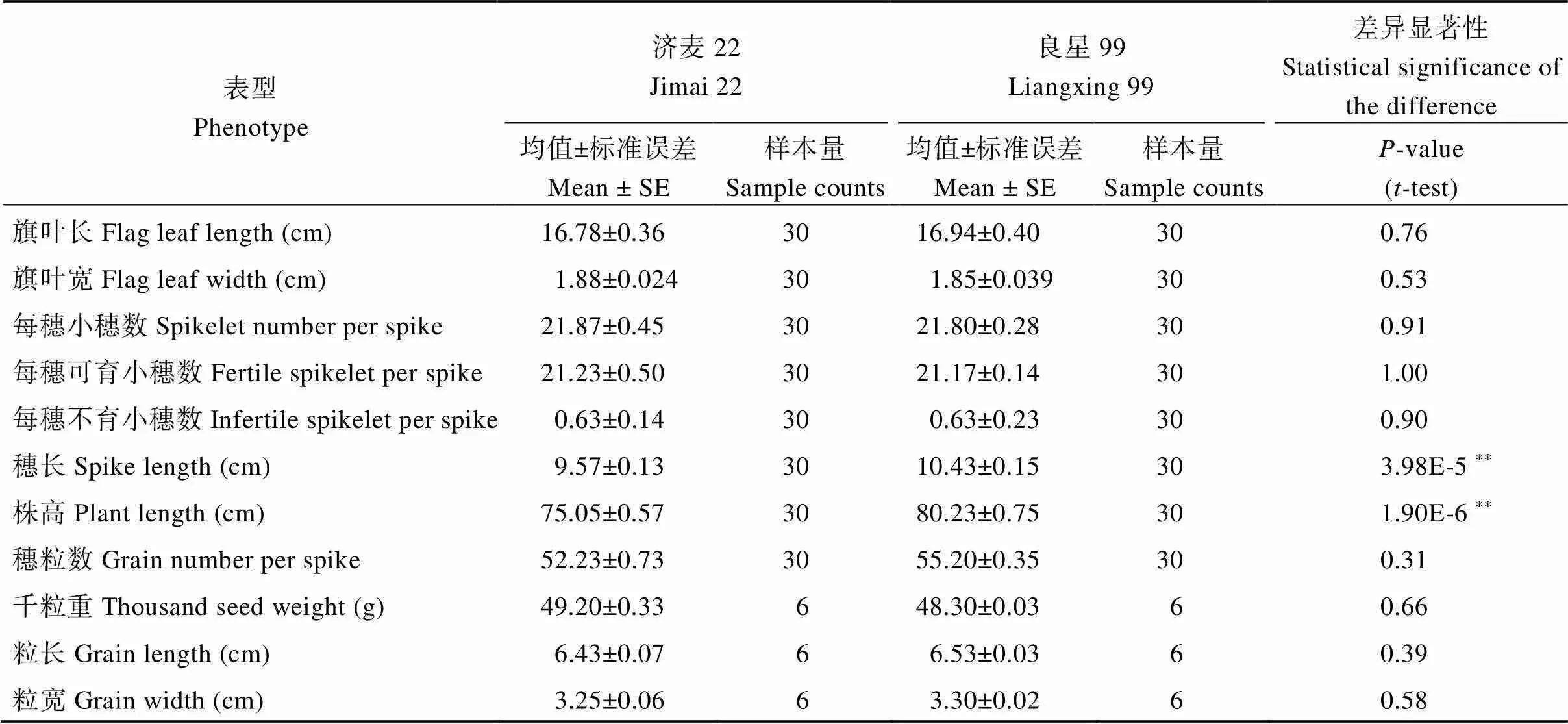
表2 济麦22与良星99的主要农艺性状差异显著性分析
**表示在0.01水平上差异显著。
**represents significantly different at the 0.01 probability level.
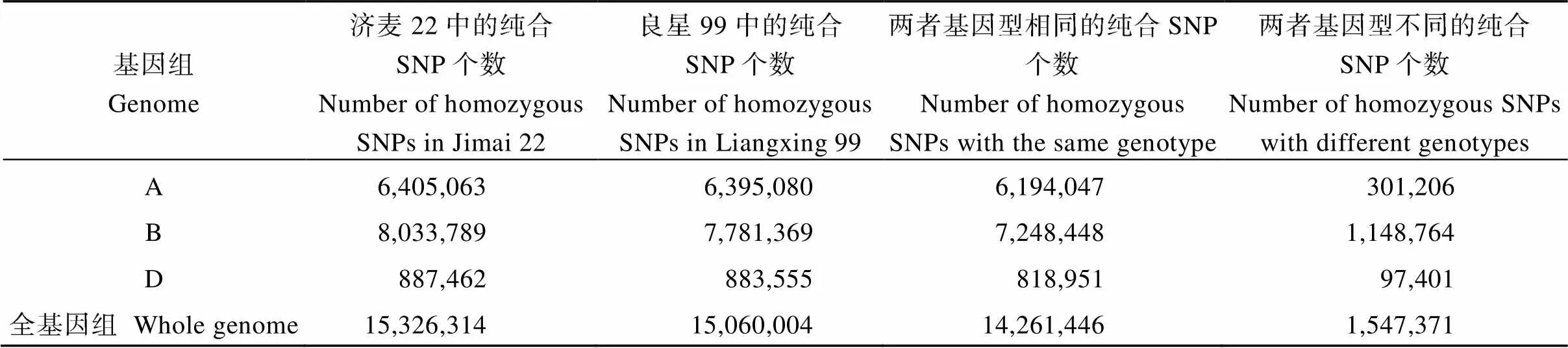
表3 济麦22与良星99的纯合SNP位点统计
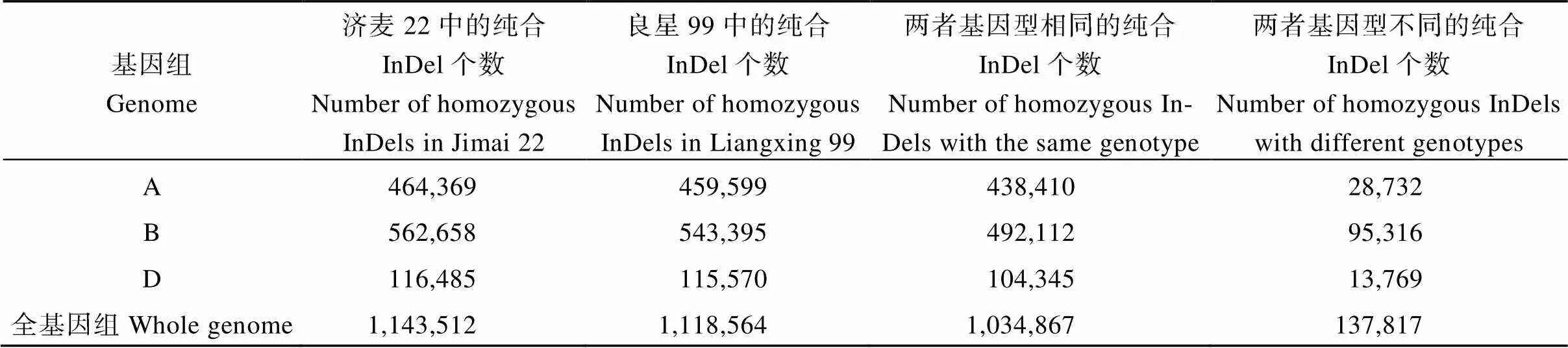
表4 济麦22与良星99的纯合InDel位点统计
2.3 济麦22与良星99的基因组CNV区间的鉴定与比较
济麦22的基因组相对于中国春参照基因组中存在557 Mb的CNV变异区间, 占全基因组的4.0%; 而在良星99基因组中检测出511 Mb的CNV变异区间, 占全基因组的3.6% (表5)。其中, 两品种共有的CNV变异区间的长度为466 Mb, 主要集中于2B、2D、5A和6A染色体; 两品种特有的CNV区间分别为91 Mb和45 Mb, 共计约占总CNV区间的22.6%, 主要集中在2B和4B染色体上(图2)。
为了验证CNV区间的分析结果, 在2B和2D染色体上的CNV差异区间中设计了特异PCR引物。其中2B_LX99_CNV1与2B_LX99_CNV2位于2B染色体上良星99特有CNV区间(Chr.2B: 667 Mb~ 726 Mb), 2B_COM_CNV位于2B染色体上良星99与济麦22共有的CNV区间(Chr. 2B: 738 Mb~769 Mb), 2D_COM_CNV位于2D染色体上济麦22与良星99共有的CNV区间(Chr. 2D: 570 Mb~638 Mb)。利用这4对引物在济麦22、良星99和中国春材料内进行PCR扩增, 结果表明材料间的差异CNV区间可以通过扩增条带的有无进行验证(图3); 其中2B_ LX99_CNV1与2B_LX99_CNV2在中国春与济麦22中均扩增出目标条带, 但未在良星99中扩增出条带, 证明该区间仅在良星99中丢失, 2B_ COM_CNV与2D_COM_CNV仅在中国春中扩增出目标条带, 证明该区间在济麦22与良星99中均丢失。PCR验证结果与全基因组分析结果吻合, 表明该分析方法计算得到的CNV变异准确可靠。
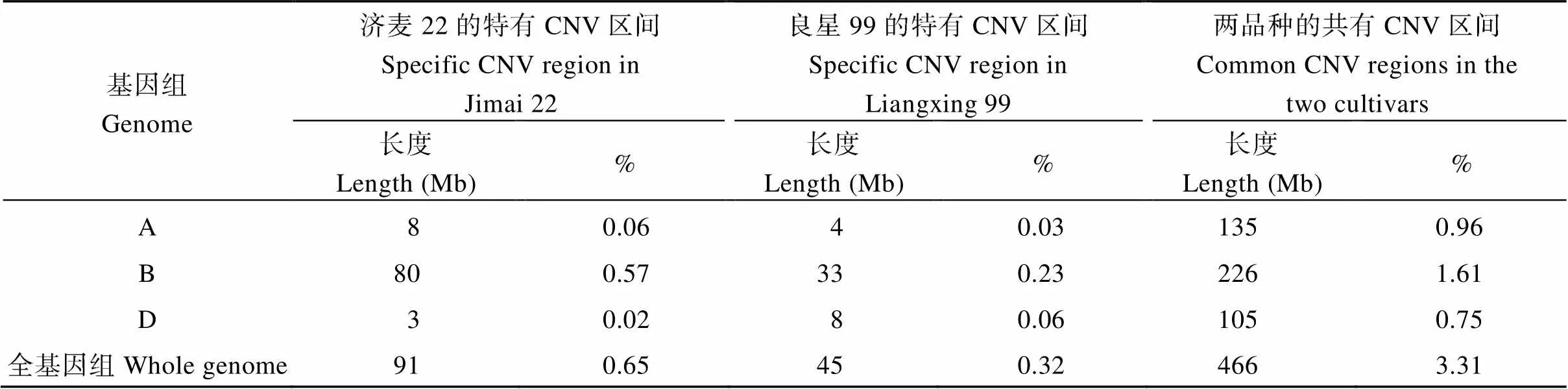
表5 济麦22与良星99的全基因组CNV区间长度统计
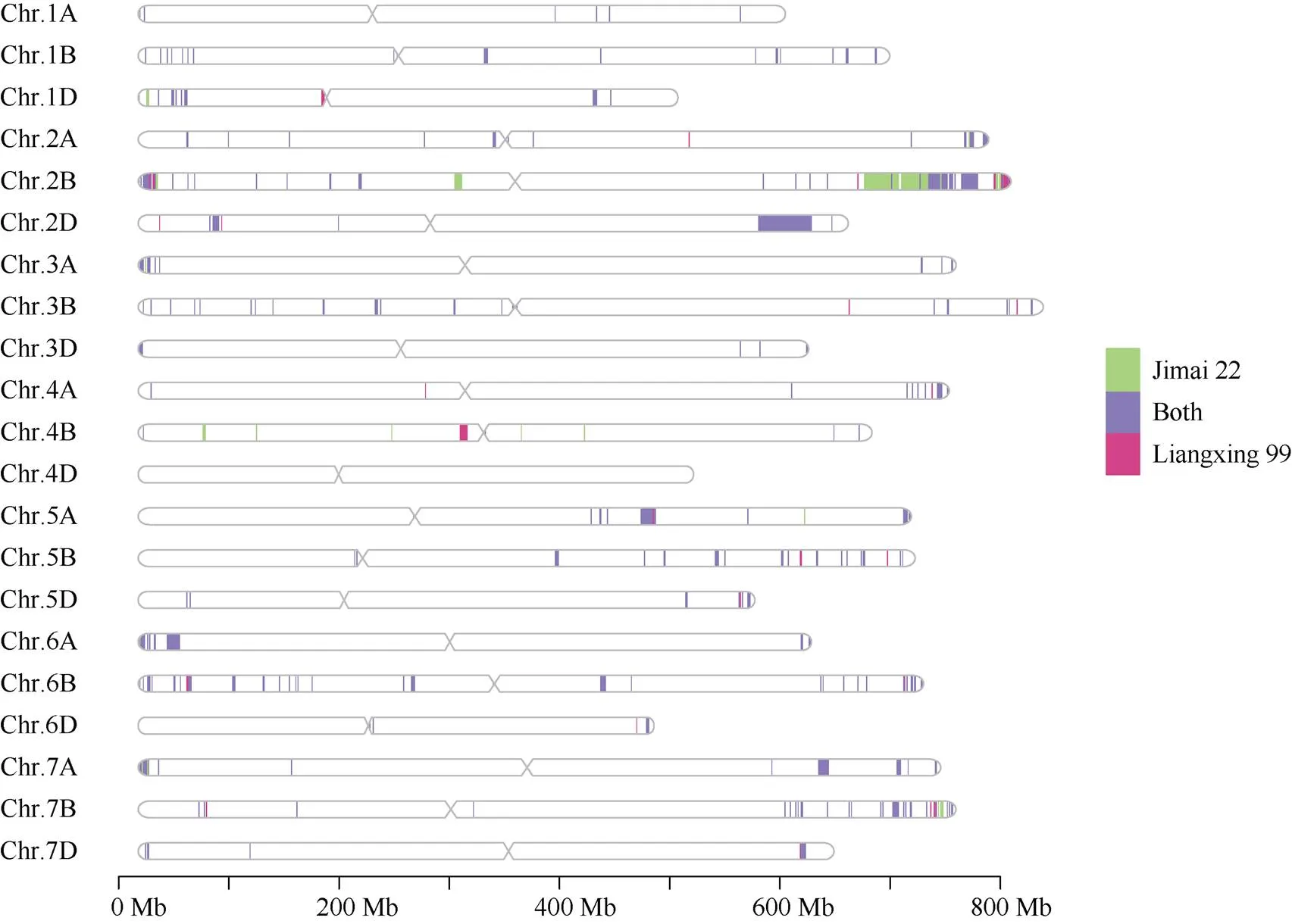
图2 济麦22与良星99的CNV区间(相对于中国春参照基因组)在各染色体上的分布
绿色: 济麦22的特有CNV区间; 粉色: 良星99的特有CNV区间; 紫色: 济麦22与良星99的共有CNV区间。
Green: specific CNV regions in Jimai 22; Pink: specific CNV regions in Liangxing 99; Purple: common CNV regions in the two varieties.
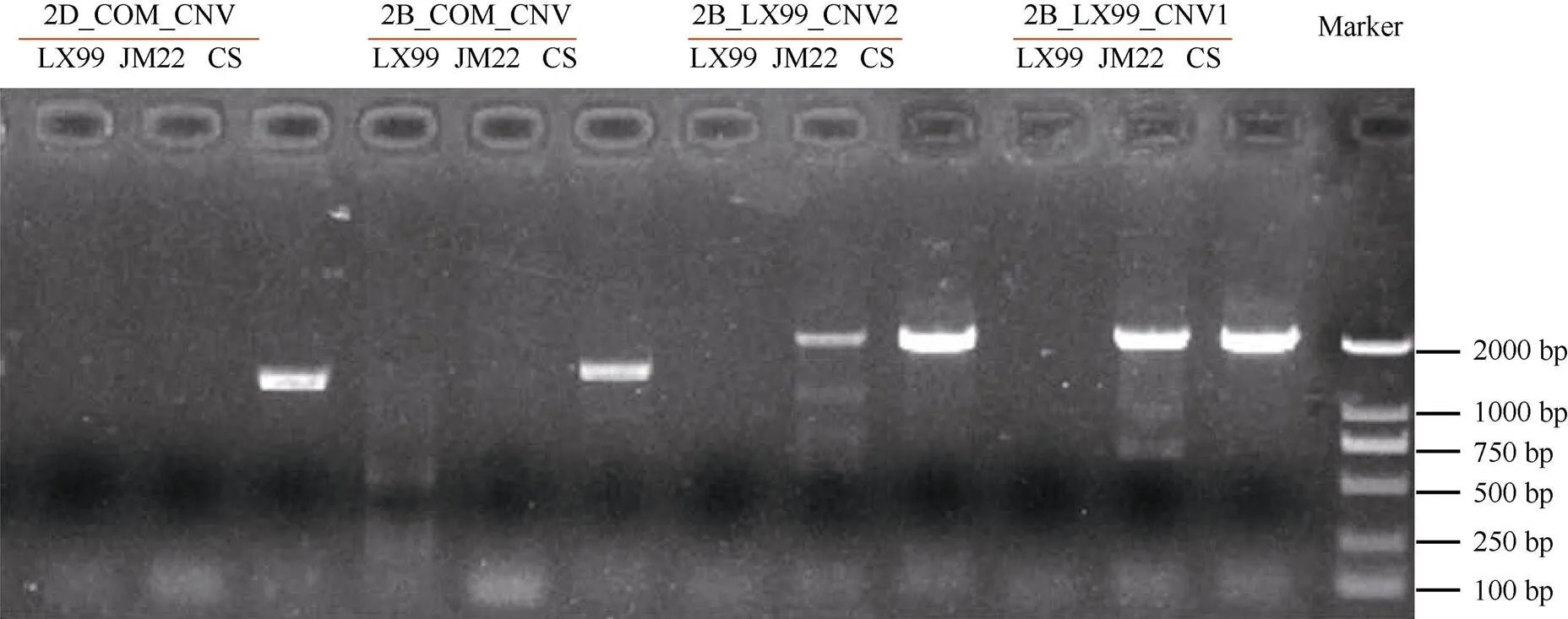
图3 利用PCR对济麦22(JM22)、良星99(LX99)和中国春(CS)中检测的CNV区间进行验证
2D_COM_CNV和2B_COM_CV分别为2D和2B染色体上共有区间序列设计的引物; 2B_LX99_CNV1和2B_LX99_CNV2为在2B染色体上良星99特有的CNV区间中序列设计的引物。每对引物对应的3个泳道从左到右分别为: 良星99(LX99)、济麦22(JM22)和中国春(CS)。
2D_COM_CNV and 2B_COM_CNV were designed utilizing sequences in common CNV regions in 2D and 2B chromosomes, respectively; 2B_LX99_CNV1 and 2B_ LX99_CNV2 were designed utilizing sequences in Liangxing 99-specific CNV regions in chromosome 2B. The corresponding three lanes from left to right for each primer are Liangxing 99 (LX99), Jimai 22 (JM 22) and Chinese Spring (CS).
通过对济麦22与良星99中特有的CNV区间内高可信度(HC)基因进行GO富集分析发现, 在济麦22的特有CNV区间内有982个基因, 其中294个基因显著富集在25个GO条目, 主要包括“ADP 结合”(GO: 0043531)、“肽酶抑制剂活性”(GO: 0030414),辅酶结合”(GO: 0050662)、“酶抑制剂活性”(GO: 0004857)、“半胱氨酸型内肽酶抑制剂活性”(GO: 0004869)等分子功能, 以及“肽酶活性负调控”(GO: 0010466)、“内肽酶活性负调控”(GO: 0010951)、“催化活性负调控”(GO: 0043086)、“碳代谢过程”(GO: 0008152)等生物学过程中, 细胞组分分类内未有富集(图4-A)。在良星99特有CNV区间内包括513个基因, 其中富集到的395个基因分布在25个GO条目中, 主要有“ADP结合”(GO: 0043531)、“血红素结合”(GO: 0020037)、“过氧化物酶活性”(GO: 0004601)、“萜烯合酶活性”(GO: 0010333)、“O-甲基转移酶活性”(GO: 0008171)、“腺苷脱氨酶活性”(GO: 0004000)等分子功能, 以及“抗氧化反应”(GO: 0006979)、“过氧化氢分解过程”(GO: 0042744)、“细胞氧化解毒”(GO: 0098869)、“蛋白磷酸化”(GO: 0006468)、“糖原生物合成”(GO: 0005978)等生物学过程和“胞外区”(GO:0005576)这一细胞组分中(图4-B)。济麦22特有CNV区间内富集在GO条目, 如“生长素反应”(GO: 0009733), “色素合成过程”(GO: 0046148)下的基因, 可能是影响两材料间的表型差异的候选基因。
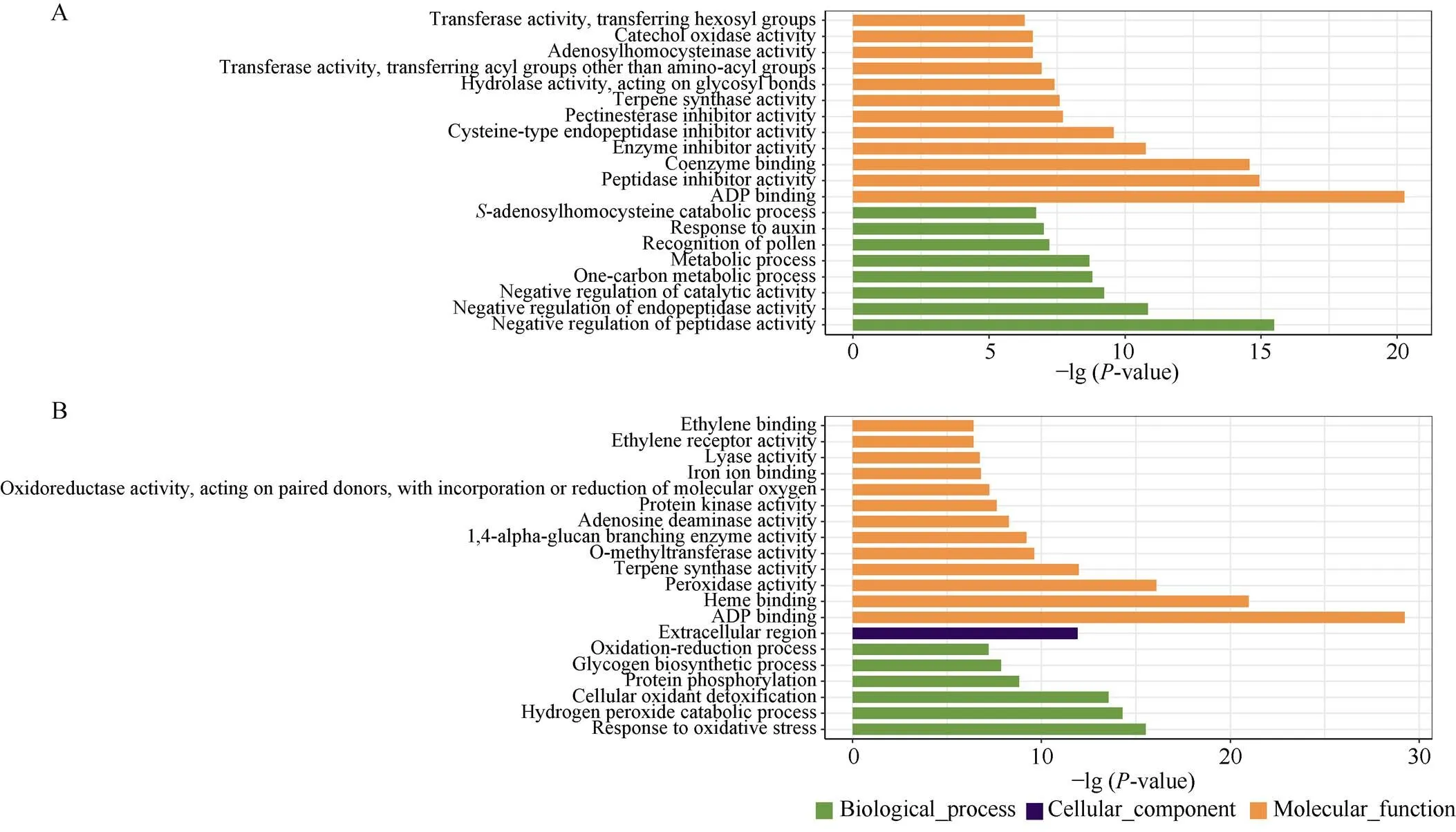
图4 济麦22(A)与良星99(B)特有CNV区间中基因的GO富集分析结果
绿色: 生物过程; 紫色: 细胞组分; 橙色: 分子功能。
2.4 济麦22和良星99间的SNP多态性热点区间和序列相似区间分析
以中国春IWGSCv1参考基因组为模板, 利用剔除品种间大片段CNV后的13.5 G大小的基因组序列进行分析。以差异纯合SNP的密度为3.1×10–5为阈值将染色体区分为“多态性热点区间”(高密度差异SNP区间)和“序列相似区间”(低密度差异SNP区间)。济麦22与良星99之间共有1915 Mb (14.2%)区间为多态性热点区间(图5-A)。在多态性热点区间中, 两品种间有1,375,981个纯合差异SNP位点, 约占两品种全部纯合差异SNP位点总数的96.5% (图5-B), 这些多态性热点区间主要集中于1D、2B、3B和4B染色体(图5-C)。
通过对两品种多态性热点区间内的14,306个高可信度基因进行GO富集分析, 其中1461个基因显著富集在20个GO条目中, 主要包括“5'–3' RNA聚合酶活性” (GO: 0003899)、“二磷酸核酮糖羧化酶活性” (GO: 0016984)、“光合作用的循环电子转运通路内转移电子的电子转运器活性” (GO: 0045156)、“醛糖1-表异构酶活性” (GO: 0004034)、以NAD或NADP为受体提供CH-OH基团的氧化还原酶活性” (GO: 0016616)等分子功能, “光合作用” (GO: 0015979)、“己糖代谢过程” (GO: 0019318)和“光系统II中的光合电子传输” (GO: 0009772)等生物过程, 以及“光系统II” (GO: 0009523)、“光系统II反应中心” (GO: 0009539)、“叶绿体” (GO: 0009507)、“质体” (GO: 0009536)和“类囊体” (GO: 0007579)等细胞组分内(图6)。综合看来, 该部分基因的GO富集分析结果主要集中在光合作用通路相关条目。
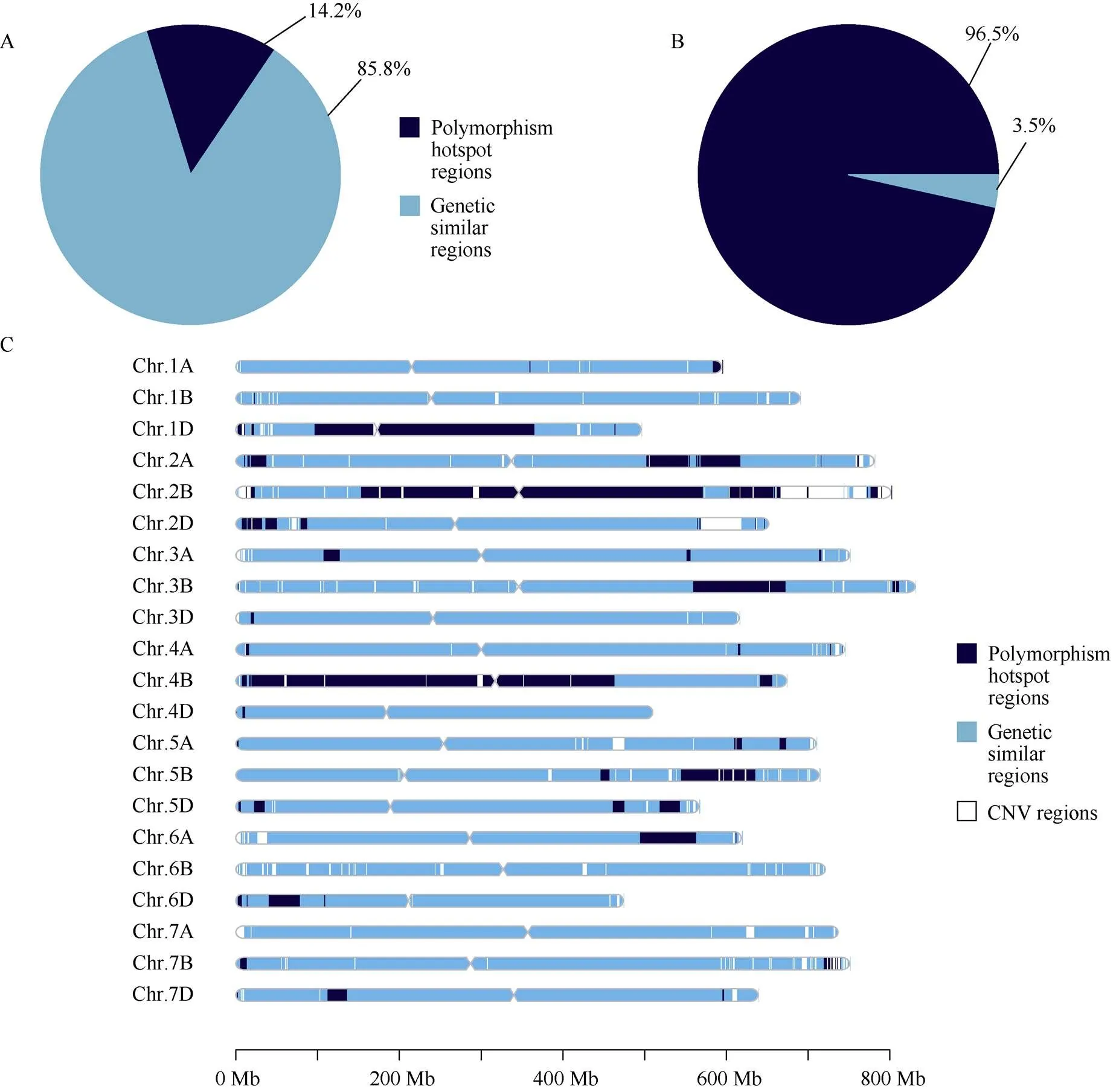
图5 济麦22与良星99全基因组内多态性热点区间分布
A: 两品种间差异SNP位点中分布在多态性热点区间与序列相似区间的比例; B: 多态性热点区间与序列相似区间的长度在全基因组上的比例; C: 多态性热点区间与序列相似区间在全基因组分布。深蓝色: 多态性热点区间; 浅蓝色: 序列相似区间; 白色: CNV区间。
A: pie chart for the proportions of differential SNPs in the polymorphism hotspot regions (PHRs) and the genetic similar regions (GSRs); B: pie chart for the proportions of the PHRs and GSRs in length; C: distributions of the PHRs and the GSRs across chromosomes.

图6 济麦22与良星99间多态性热点区间的基因GO富集分析结果
绿色: 生物过程; 紫色: 细胞组分; 橙色: 分子功能。
2.5 济麦22和良星99的多态性热点区间和序列相似区间的突变类型分析
为比较两品种间多态性热点区间和序列相似区间中的突变类型的异同, 我们首先统计差异SNP位点的碱基变异类型的频率(图7-A和表6)。无论在多态性热点区间还是序列相似区间, T↔C和G↔A类型的突变频率均为最多, 在多态性热点区间内约占71.4%, 略高于在序列相似区间内所占总SNP的比例(66.5%)。同时, 我们还统计了两品种间差异InDel的长度分布(图7-B和表7)。多态性热点区间中的单碱基的InDel占该区间内所有InDel位点的64.7%, 其比例高于单碱基的InDel在序列相似区间中所占的比例(49.0%)。
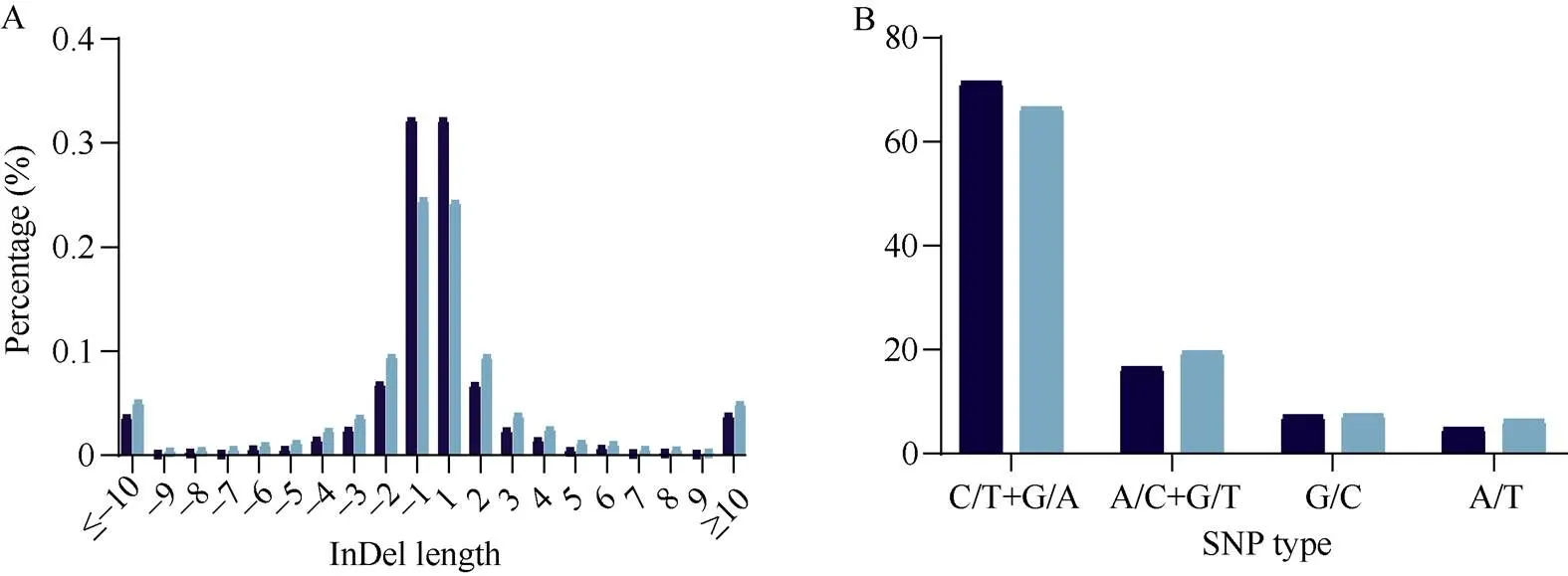
图7 济麦22与良星99间多态性热点区间与序列相似区间中各自的InDel长度分布(A)和SNP突变类型(B)
深蓝: 多态性热点区间; 浅蓝: 序列相似区间。
Dark blue: polymorphism hotspot regions; Light blue: genetic similar regions.
我们分别对多态性热点区间与序列相似区间内两品种间差异SNP与InDel位点对于基因功能的影响进行了注释与比较(表8), 结果表明多态性热点区间与序列相似区间中基因序列上的SNP和InDel变异的类型主要以错义突变(missense variation)和同义突变(synonymous variation)为主。在多态性热点区间中错义突变(51.3%)与终止子相关突变(stop-codon gain/lost)(1.5%)所占比例要分别略高于两者在序列相似区间的44.7%和0.8%比例。
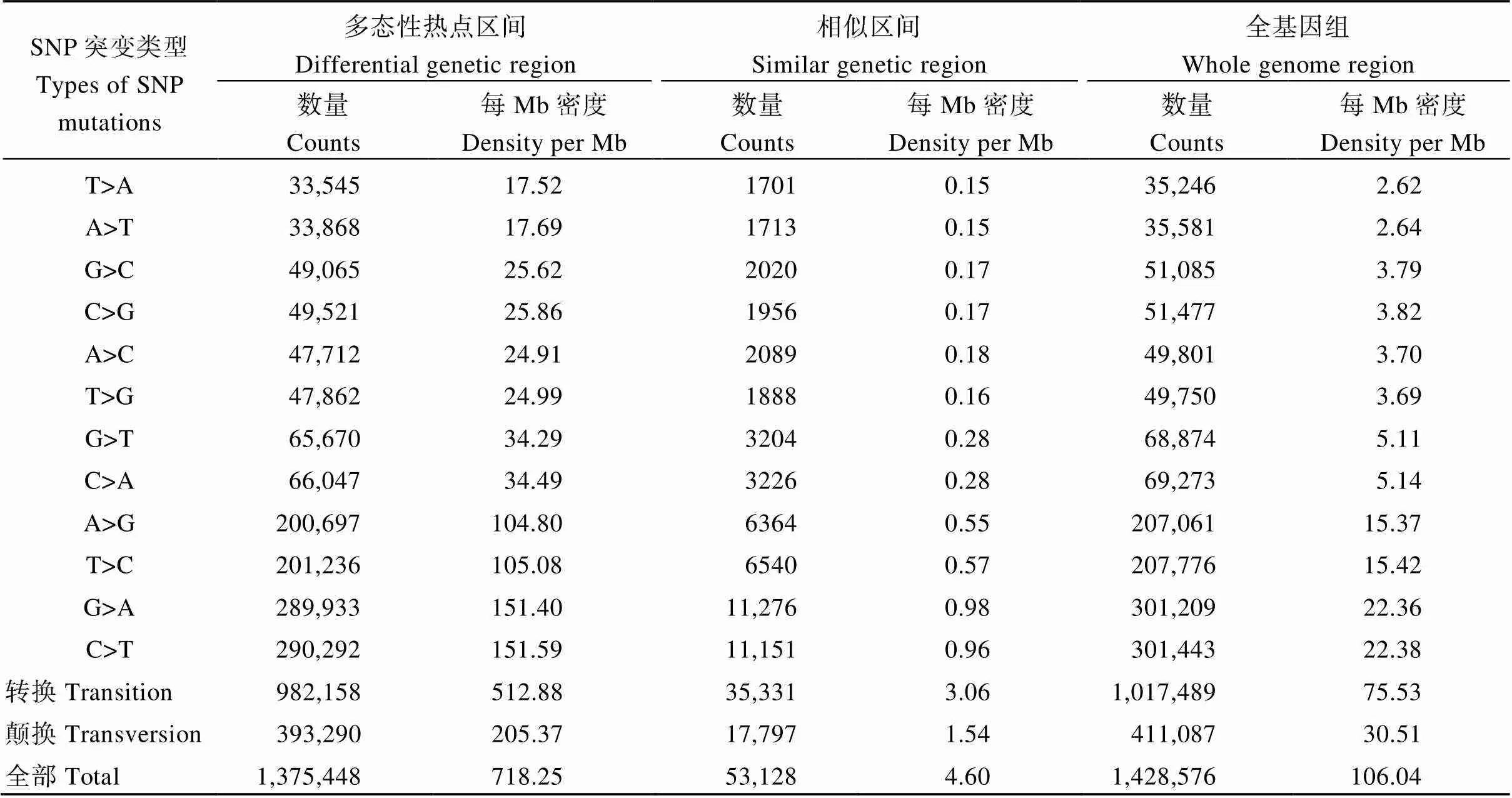
表6 济麦22与良星99全基因组差异纯合SNP的突变类型的统计
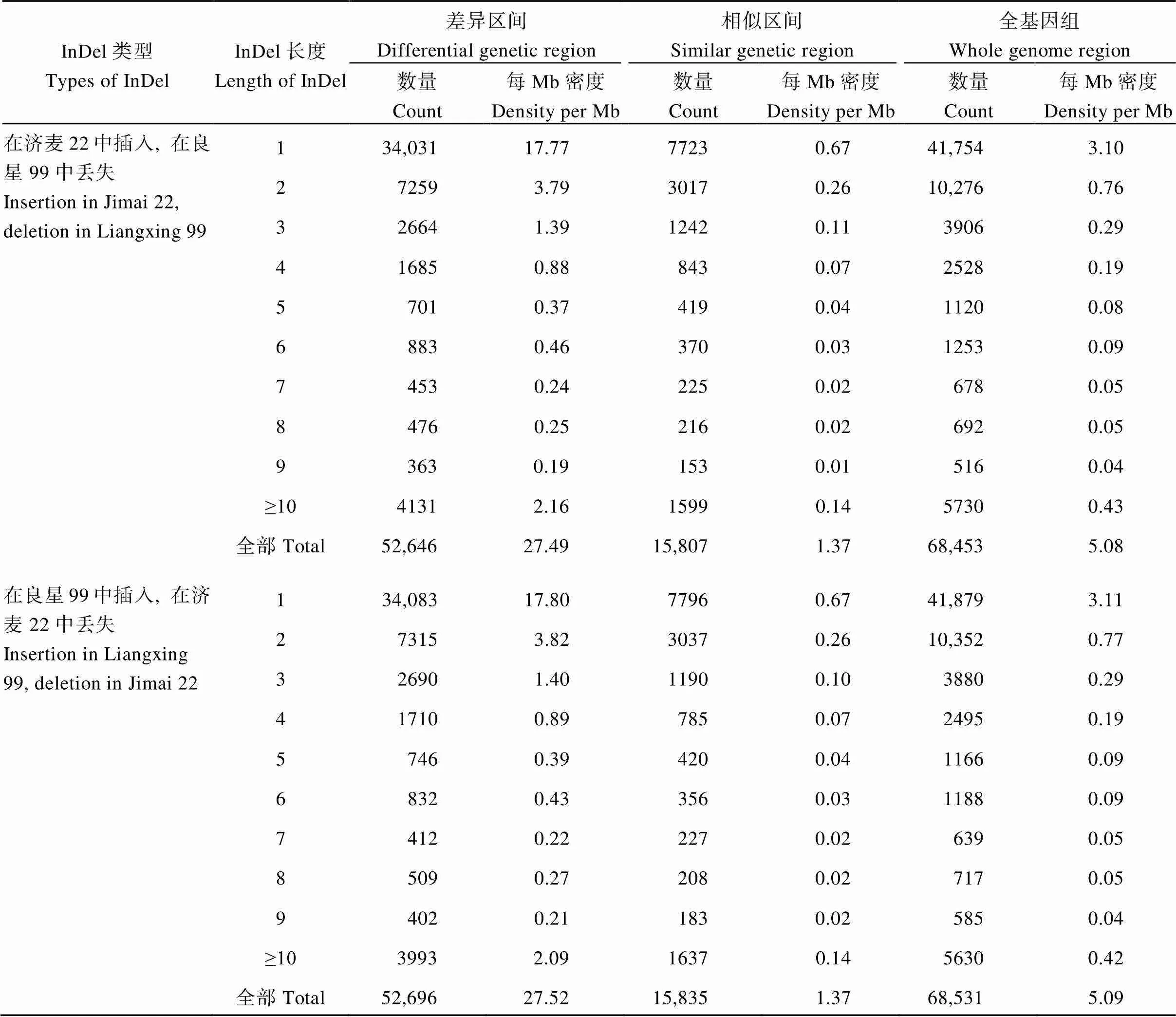
表7 济麦22与良星99的纯合InDel变异的长度分布
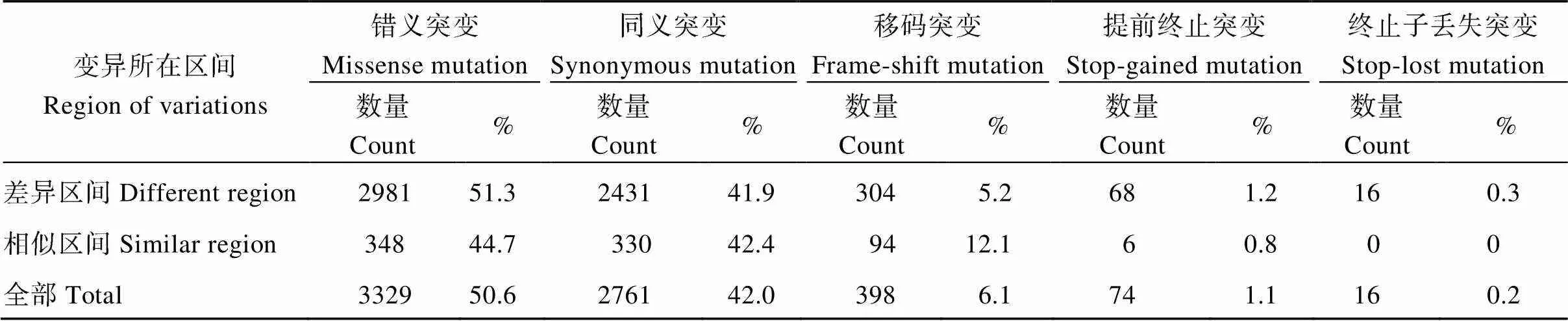
表8 济麦22与良星99的纯合点突变对蛋白编码功能影响的统计
2.6 株高候选基因在济麦22和良星99之间的序列差异分析及重测序结果验证
济麦22与良星99在株高和穗长上存在显著差异, 利用两者的重测序数据对小麦目前已克隆的矮秆基因[18]、[18]、株高相关基因[19]和在2D染色体的内定位到2个株高穗长候选基因[20]等进行序列分析, 发现区间内的2个基因在品种间存在序列差异(表9)。
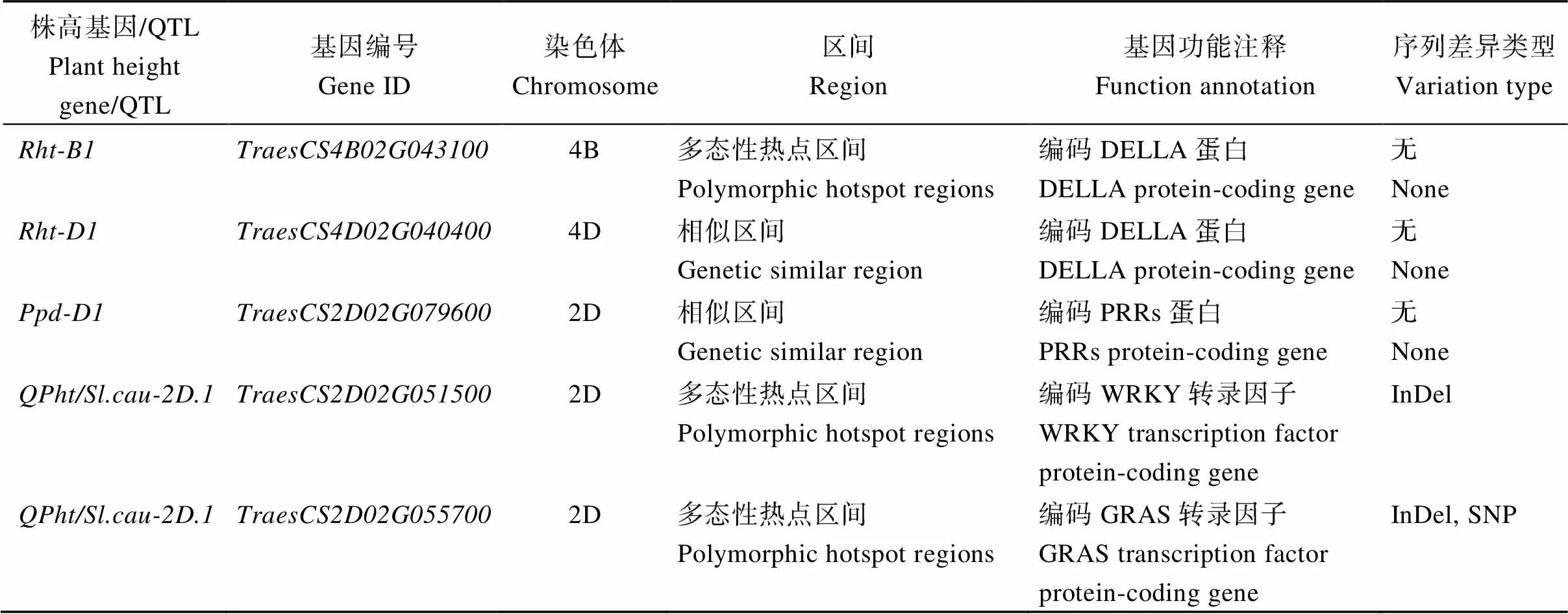
表9 小麦株高相关基因列表及在序列相似区间和多态性热点区间的分布
利用基因特异性引物对QPht/Sl.cau-2D.1 QTL区间内的和基因分别在济麦22与良星99中进行扩增、测序发现, 2个基因在品种间存在引起氨基酸改变的差异位点(图8)。该QTL区间是矮秆基因所在区间, 该位点对小麦的株高和穗长均有影响[21], 但其遗传功能仍需进一步试验验证。
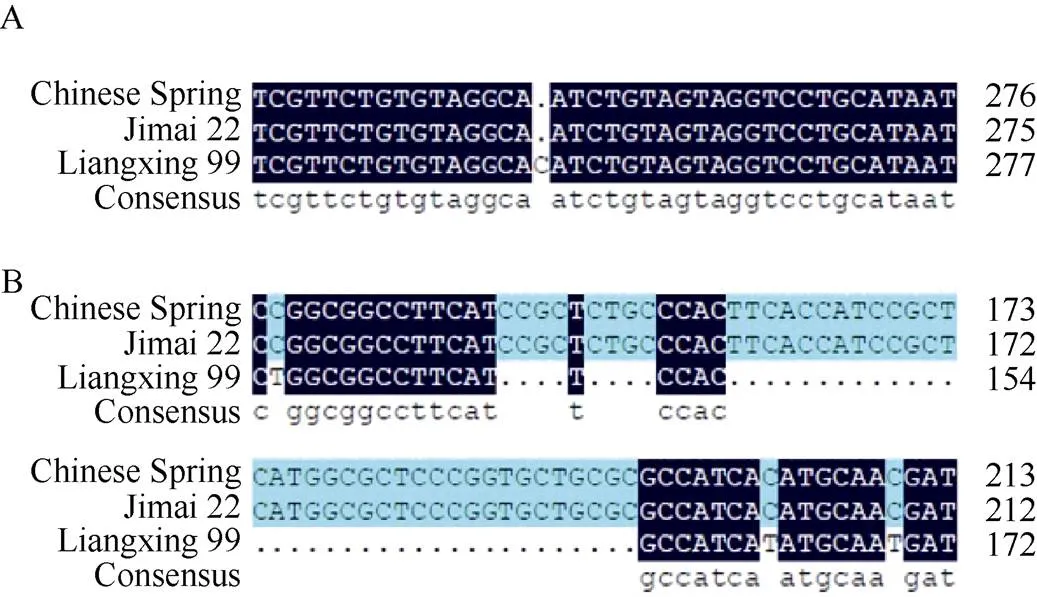
图8 TraesCS2D02G051500(A)与TraesCS2D02G055700(B)在中国春、济麦22和良星99中功能改变位点附近的核苷酸序列
3 讨论
本研究通过对济麦22和良星99两个小麦品种的重测序数据进行分析, 系统研究了两个品种在基因组水平上存在的CNV区间和SNP热点区间。与中国春参照基因组比较发现, 两个品种均有大量CNV丢失变异区间, 约占全基因组的4.3%。除集中分布在2B染色体上148 Mb的CNV变异外, 大部分CNV变异区间零散地分布在不同染色体上。这一结果表明, 与中国春参考基因组序列相比, 济麦22和良星99中存在大量的CNV变异(特别是CNV丢失变异)。通过比较2个材料中CNV变异区间的异同, 发现大部分CNV区间是共有的, 但仍有22.6%的差异CNV区间; 其中60.3%的差异CNV区间集中于2B染色体。需要指出的是, 本研究中发现的“CNV缺失区间”是通过读段比对后的覆盖度确定的, 不能排除该区域可能来源于遗传距离较远的基因组序列的可能性。根据品种间差异SNP的分布密度对染色体区间进行了划分, 鉴定出基因组14.2%的区间是两品种间的多态性热点区间, 集中了全基因组96.5%差异SNP位点, 这些区间主要位于1D、2B和4B染色体上。通过鉴定相似区间和多态性热点区间变异位点的特征, 发现两类区间在SNP变异类型、InDel长度分布、突变影响编码功能的比例上有一定区别。上述结果表明济麦22和良星99在全基因组水平具有较高的相似性, 但在特定的染色体上确实存大片段的差异区间。
济麦22和良星99的主要农艺和产量性状存在较高相似性, 只有在株高和穗长上存在差异。结合品种间序列差异位点分析, 我们推测控制品种间株高和穗长的差异基因可能位于差异SNP热点区域或差异CNV区间。结合目前已经在小麦中克隆的株高或穗长的5个相关基因, 并分析两品种间基因序列, 发现了2个位于差异SNP热点区且存在影响编码功能的变异的候选基因。本研究在已有基因组变异和表型信息的基础上对已知性状相关基因和品种间差异序列区间进行了初步分析, 找到了位于多态性热点区间的2个具有序列差异的基因, 但其生物学功能和关联仍需进一步验证。利用表型差异和全基因组序列多态性分析相结合的方法, 本研究的方法可为今后借助基因组测序方法快速定位和克隆候选基因提供了新的参考。
Zhao等[22]研究发现, 良星99的2BL上有一个控制抗白粉病的主效基因, 该基因被命名为[23]。利用中国春参照基因组的信息, 将基因定位在chr2B的581 Mb~596 Mb的区间[24]。本研究的结果显示, 济麦22和良星99的2BL的绝大部分区间为多态性热点区间, 但575 Mb~603 Mb区段为本研究中鉴定的“序列相似区间”, 该区段包含所在的物理区间; 本研究间接支持了Qu等[24]在济麦22和良星99的衍生材料中均携带相同的基因的结论。该结果也表明本研究鉴定的序列相似区间可为抗病等关键性状基因的定位研究提供参考。
小麦品种的重测序研究为探索小麦基因组序列的多态性提供高维度、高精细度、类型丰富的基因组变异信息。本研究也为比较小麦品种间的基因组序列差异分析提供了方法学参考。随着小麦材料重测序数据的不断积累, 小麦品种间基因组序列变异的特征和规律也将得到进一步揭示。有效分析和利用丰富的基因组变异信息, 将有助于对关键性状基因功能进行阐释, 辅助小麦育种对优异基因区间的利用, 提高小麦品种选育的效率。
4 结论
济麦22和良星99两个重要小麦品种的田间表型相似。本研究通过对两个品种进行全基因序列差异分析发现, 济麦22和良星99间的遗传组成相似性较高(序列相似区间占85.8%), 但仍存在大区段CNV变异(136 M)和占全基因组14.2%的差异多态性热点区间; 其中差异SNP热点区间主要集中在1D、2B和4B染色体上。本工作为进一步研究和利用济麦22和良星99提供了重要的基因组变异数据; 也为小麦品种间的基因组变异热点区间的鉴定提供了方法参考。
说明: 本研究中的原始测序数据已提交至中国科学院北京基因组研究所BIG数据中心[25]的组学原始数据归档库[26]中(https://bigd.big.ac.cn/gsa), 编号为CRA002333。为便于数据的共享, 我们利用SnpHub数据库模型[27]搭建了2个材料的基因组变异信息查询数据库(http://wheat.cau.edu.cn/Wheat_SnpHub_ Portal/lx99_jm22/)。
[1] 何中虎, 庄巧生, 程顺和, 于振文, 赵振东, 刘旭.中国小麦产业发展与科技进步. 农学学报, 2018, 8(1): 99–106. He Z H, Zhuang Q S, Cheng S H, Yu Z W, Zhao Z D, Liu X. Wheat production and technology improvement in China., 2018, 8(1): 99–106 (in Chinese).
[2] 张婷, 逯腊虎, 张伟, 杨斌, 袁凯, 史晓芳. 黄淮麦区300个小麦种质的主要农艺性状比较.小麦研究, 2018, 39(1): 13–20. Zhang T, Lu L H, Zhang W, Yang B, Yuan K, Shi X F. Comparison of main agronomic traits of 300 wheat germplasms in Huanghuai Wheat Region., 2018, 39(1): 13–20 (in Chinese).
[3] 曹廷杰, 王西成, 赵虹. 国审小麦新品种周麦18号丰产性、稳产性及适应性分析. 中国农业科技导报, 2007, 9(1): 39–41. Cao T J, Wang X C, Zhao H. Analysis on the yield potential, stability and adaptability of national authorized wheat variety Zhoumai 18., 2007, 9(1): 39–41 (in Chinese with English abstract)
[4] 盖红梅, 李玉刚, 王瑞英, 李振清, 王圣健, 高峻岭, 张学勇. 鲁麦14对山东新选育小麦品种的遗传贡献. 作物学报, 2012, 38: 954–961. Gai H M, Li Y G, Wang R Y, Li Z Q, Wang S J, Gao J L, Zhang X Y. Genetic contribution of Lumai 14 to novel wheat varieties developed in Shandong province., 2012, 38: 954–961 (in Chinese with English abstract)
[5] 傅晓艺, 郭进考, 刘艳滨, 史占良, 单子龙, 韩然, 何明琦. 利用石4185为骨干亲本培育高产小麦新品种. 种子, 2017, 36(12): 95–99. Fu X Y, Guo J K, Liu Y B, Shi Z L, Shan Z L, Han R, He M Q. New wheat varieties with high yield of core parent breeding by using Shi 4185., 2017, 36(12): 95–99 (in Chinese).
[6] 凌宏清. 小麦及其近缘种基因组测序研究进展与发展趋势. 麦类作物学报, 2016, 36: 397–403. Ling H Q. Progress and perspectives of the genome sequencing in wheat and its relatives., 2016, 36: 397–403 (in Chinese with English abstract)
[7] Uauy C. Wheat genomics comes of age., 2017, 36: 142–148.
[8] 殷贵鸿, 李根英, 何中虎, 刘建军, 王辉, 夏先春. 小麦新品种济麦22抗白粉病基因的分子标记定位. 作物学报, 2009, 35: 1425–1431. Yin H G, Li G Y, He Z H, Liu J J, Wang H, Xia X C. Molecular mapping of powdery mildew resistance gene in wheat cultivar Jimai 22., 2009, 35: 1425–1431 (in Chinese with English abstract)
[9] 赵广军, 张英肖. 高产稳产冬小麦品种良星99栽培技术. 小麦研究, 2006, 27(3): 12–13. Zhao G J, Zhang Y X. Cultivation technique of Liangxing 99, a winter wheat variety with high and stable yield., 2006, 27(3): 12–13 (in Chinese)
[10] Qu Y F, Wu P P, Hu J H, Chen Y X, Shi Z L, Qiu D, Li Y H, Zhang H J, Zhou Y, Yang L, Liu H W, Zhu T Q, Liu Z Y, Zhang Y M, Li H J. Molecular detection of the powdery mildew resistance genes in winter wheats DH51302 and Shimai 26., 2020, 19: 931–940.
[11] Bolger A M, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data., 2014, 30: 2114–2120.
[12] Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform., 2009, 25: 1754–1760.
[13] Appels R, Eversole K, Feuillet C, Keller B, Rogers J, Stein N, Pozniak C J, Stein N, Choulet F, Distelfeld A, Eversole K, Poland J, Rogers J, Ronen G, Sharpe A G, Pozniak C, Ronen G, Stein N, Barad O, Baruch K, Choulet F, Keeble-Gagnere G, Mascher M, Sharpe A G, Ben-Zvi G, Josselin A A, Stein N, Mascher M, Himmelbach A, Choulet F, Keeble-Gagnere G, Mascher M, Rogers J, Balfourier F, Gutierrez-Gonzalez J, Hayden M, Josselin A A, Koh C, Muehlbauer G, Pasam R K, Paux E, Pozniak C J, Rigault P, Sharpe A G, Tibbits J, Tiwari V, Choulet F, Keeble-Gagnere G, Mascher M, Josselin A A, Rogers J, Spannagl M, Choulet F, Lang D, Gundlach H, Haberer G, Keeble-Gagnere G, Mayer KFX, Ormanbekova D, Paux E, Prade V, Simkova H, Wicker T, Choulet F, Spannagl M, Swarbreck D, Rimbert H, Felder M, Guilhot N, Gundlach H, Haberer G, Kaithakottil G, Keilwagen J, Lang D, Leroy P, Lux T, Mayer KFX, Twardziok S, Venturini L, Appels R, Rimbert H, Choulet F, Juhasz A, Keeble-Gagnere G, Choulet F, Spannagl M, Lang D, Abrouk M, Haberer G, Keeble-Gagnere G, Mayer KFX, Wicker T, Choulet F, Wicker T, Gundlach H, Lang D, Spannagl M, Lang D, Spannagl M, Appels R, Fischer I, Uauy C, Borrill P, Ramirez-Gonzalez R H, Appels R, Arnaud D, Chalabi S, Chalhoub B, Choulet F, Cory A, Datla R, Davey M W, Hayden M, Jacobs J, Lang D, Robinson S J, Spannagl M, Steuernagel B, Tibbits J, Tiwari V, van Ex F, Wulff BBH, Pozniak C J, Robinson S J, Sharpe A G, Cory A, Benhamed M, Paux E, Bendahmane A, Concia L, Latrasse D, Rogers J, Jacobs J, Alaux M, Appels R, Bartos J, Bellec A, Berges H, Dolezel J, Feuillet C, Frenkel Z, Gill B, Korol A, Letellier T, Olsen O A, Simkova H, Singh K, Valarik M, van der Vossen E, Vautrin S, Weining S, Korol A, Frenkel Z, Fahima T, Glikson V, Raats D, Rogers J, Tiwari V, Gill B, Paux E, Poland J, Dolezel J, Cihalikova J, Simkova H, Toegelova H, Vrana J, Sourdille P, Darrier B, Appels R, Spannagl M, Lang D, Fischer I, Ormanbekova D, Prade V, Barabaschi D, Cattivelli L, Hernandez P, Galvez S, Budak H, Steuernagel B, Jones J D G, Witek K, Wulff B B H, Yu G, Small I, Melonek J, Zhou R, Juhasz A, Belova T, Appels R, Olsen O A, Kanyuka K, King R, Nilsen K, Walkowiak S, Pozniak C J, Cuthbert R, Datla R, Knox R, Wiebe K, Xiang D, Rohde A, Golds T, Dolezel J, Cizkova J, Tibbits J, Budak H, Akpinar B A, Biyiklioglu S, Muehlbauer G, Poland J, Gao L, Gutierrez-Gonzalez J, N'Daiye A, Dolezel J, Simkova H, Cihalikova J, Kubalakova M, Safar J, Vrana J, Berges H, Bellec A, Vautrin S, Alaux M, Alfama F, Adam-Blondon A F, Flores R, Guerche C, Letellier T, Loaec M, Quesneville H, Pozniak C J, Sharpe A G, Walkowiak S, Budak H, Condie J, Ens J, Koh C, Maclachlan R, Tan Y, Wicker T, Choulet F, Paux E, Alberti A, Aury J M, Balfourier F, Barbe V, Couloux A, Cruaud C, Labadie K, Mangenot S, Wincker P, Gill B, Kaur G, Luo M, Sehgal S, Singh K, Chhuneja P, Gupta O P, Jindal S, Kaur P, Malik P, Sharma P, Yadav B, Singh N K, Khurana J, Chaudhary C, Khurana P, Kumar V, Mahato A, Mathur S, Sevanthi A, Sharma N, Tomar R S, Rogers J, Jacobs J, Alaux M, Bellec A, Berges H, Dolezel J, Feuillet C, Frenkel Z, Gill B, Korol A, van der Vossen E, Vautrin S, Gill B, Kaur G, Luo M, Sehgal S, Bartos J, Holusova K, Plihal O, Clark M D, Heavens D, Kettleborough G, Wright J, Valarik M, Abrouk M, Balcarkova B, Holusova K, Hu Y, Luo M, Salina E, Ravin N, Skryabin K, Beletsky A, Kadnikov V, Mardanov A, Nesterov M, Rakitin A, Sergeeva E, Handa H, Kanamori H, Katagiri S, Kobayashi F, Nasuda S, Tanaka T, Wu J, Appels R, Hayden M, Keeble-Gagnere G, Rigault P, Tibbits J, Olsen O A, Belova T, Cattonaro F, Jiumeng M, Kugler K, Mayer K F X, Pfeifer M, Sandve S, Xun X, Zhan B, Simkova H, Abrouk M, Batley J, Bayer P E, Edwards D, Hayashi S, Toegelova H, Tulpova Z, Visendi P, Weining S, Cui L, Du X, Feng K, Nie X, Tong W, Wang L, Borrill P, Gundlach H, Galvez S, Kaithakottil G, Lang D, Lux T, Mascher M, Ormanbekova D, Prade V, Ramirez-Gonzalez R H, Spannagl M, Stein N, Uauy C, Venturini L, Stein N, Appels R, Eversole K, Rogers J, Borrill P, Cattivelli L, Choulet F, Hernandez P, Kanyuka K, Lang D, Mascher M, Nilsen K, Paux E, Pozniak C J, Ramirez-Gonzalez R H, Simkova H, Small I, Spannagl M, Swarbreck D, Uauy C. Shifting the limits in wheat research and breeding using a fully annotated reference genome., 2018, 361: eaar7191.
[14] Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools., 2009, 25: 2078–2079.
[15] van der Auwera G A, Carneiro M O, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella K V, Altshuler D, Gabriel S, Depristo M A. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline., 2013, 43: 10–11.
[16] Cingolani P, Platts A, Wang L L, Coon M, Nguyen T, Wang L, Land S J, Lu X, Ruden D M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff., 2014, 6: 80–92.
[17] Quinlan A R, Hall I M. BEDTools: a flexible suite of utilities for comparing genomic features., 2010, 26: 841–842.
[18] Peng J, Richards D E, Hartley N M, Murphy G P, Devos K M, Flintham J E, Beales J, Fish L J, Worland A J, Pelica F, Sudhakar D, Christou P, Snape J W, Gale M D, Harberd N P. ‘Green revolution’ genes encode mutant gibberellin response modulators., 1999, 400: 256–261.
[19] Beales J, Turner A, Griffiths S, Snape J W, Laurie D A. A pseudo-response regulator is misexpressed in the photoperiod insensitivemutant of wheat (L.)., 2007, 115: 721–733.
[20] 柴岭岭. 普通小麦2D染色体株高和穗长QTL的精细定位与图位克隆. 中国农业大学博士学位论文, 北京, 2019. pp 38–39. Chai L L. Fine Mapping and Map-based Cloning of QTL Controlling Plant Height and Spike Length on Chromosome 2D in Common Wheat (L.). PhD Dissertation of China Agricultural University, Beijing, China, 2019. pp 38–39 (in Chinese with English abstract).
[21] Chai L L, Chen Z Y, Bian R L, Zhai H J, Cheng X J, Peng H R, Yao Y Y, Hu Z R, Xin M M, Guo W L, Sun Q X, Zhao A J, Ni Z F. Dissection of two quantitative trait loci with pleiotropic effects on plant height and spike length linked in coupling phase on the short arm of chromosome 2D of common wheat (L.).,2018, 131: 2621–2637.
[22] Zhao Z H, Sun H G, Song W, Lu M, Huang J, Wu L F, Wang X M, Li H J. Genetic analysis and detection of the gene MlLX99 on chromosome 2BL conferring resistance to powdery mildew in the wheat cultivar Liangxing 99., 2013, 126: 3081–3089.
[23] 邹景伟, 邱丹, 孙艳玲, 郑超星, 李静婷, 吴培培, 武小菲, 王晓鸣, 周阳, 李洪杰.——小麦品种良星99抗白粉病基因的有效性. 作物学报, 2017, 43: 332–342.Zou J W, Qiu D, Sun Y L, Zheng C X, Li J T, Wu P P, Wu X F, Wang X M, Zhou Y, Li H J.: effectiveness of the gene conferring resistance to powdery mildew in wheat cultivar Liangxing 99., 2017, 43: 332–342 (in Chinese with English abstract).
[24] Wu P P, Hu J H, Zou J W, Qiu D, Qu Y F, Li Y H, Li T, Zhang H J, Yang L, Liu H W, Zhou Y, Zhang Z J, Li, J T, Liu Z Y, Li H J. Fine mapping of the wheat powdery mildew resistance geneusing comparative genomics analysis and the Chinese Spring reference genomic sequence., 2019, 132: 1451–1461.
[25] Wang Y, Song F, Zhu J, Zhang S, Yang Y, Chen T, Tang B, Dong L, Ding N, Zhang Q, Bai Z, Dong X, Chen H, Sun M, Zhai S, Sun Y, Yu L, Lan L, Xiao J, Fang X, Lei H, Zhang Z, Zhao W. GSA: genome sequence archive., 2017, 15: 14–18.
[26] National genomics data center members and partners., 2020, 48: D24–D33.
[27] Wang W X, Wang Z H, Li X T, Ni Z F, Hu Z R, Xin M M, Peng H R, Yao Y Y, Sun Q X, Guo W L. SnpHub: an easy-to-set-up web server framework for exploring large-scale genomic variation data in the post-genomic era with applications in wheat., 2020, 9: giaa060.
附图1 济麦22(A)与良星99(B)全基因组reads覆盖度的密度分布直方图
X轴为经归一化的每1 Mb内的平均reads覆盖度; Y轴为覆盖度的密度。
X-axis, the averaged read coverage per 1 Mb after normalization. Y-axis, the distribution density. A and B represent density plot of bin-wise normalized average read coverage in Jimai 22 and Liangxing 99, respectively.
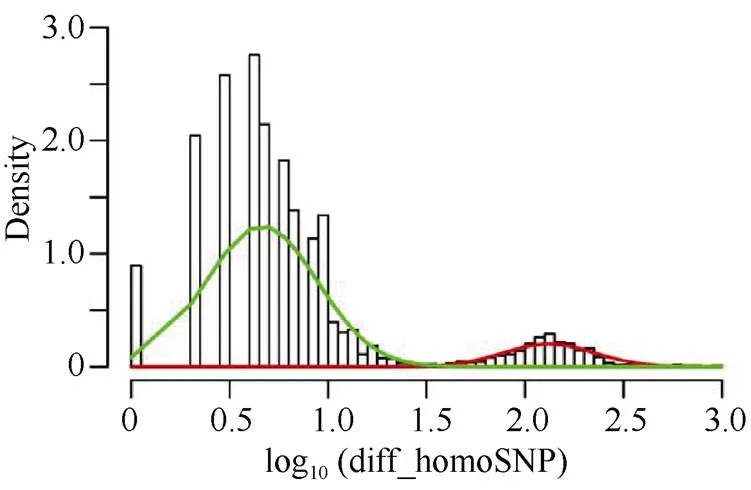
附图2 济麦22与良星99间的单区间差异SNP的密度分布图
X轴, 每Mb区间内差异SNP位点数的对数(以10为底); Y轴, 位点数的密度。
X-axis, 10-based logarithm of the counts of differential SNP sites per Mb. Y-axis, the distribution density.
Comparative analysis of the genomic sequences between commercial wheat varieties Jimai 22 and Liangxing 99
YANG Zheng-Zhao, WANG Zi-Hao, HU Zhao-Rong, XIN Ming-Ming, YAO Ying-Yin, PENG Hui-Ru, YOU Ming-Shan, SU Zhen-Qi*, and GUO Wei-Long*
College of Agronomy and Biotechnology / State Key Laboratory for Agrobiotechnology / Key Laboratory of Crop Heterosis and Utilization, Ministry of Education, China Agricultural University, Beijing 100193, China
Jimai 22 and Liangxing 99 are high-yield wheat varieties widely planted in the North Huang-Huai Rivers Valley Winter Wheat Zone and Northern Winter Wheat Zone in China, and are currently used as important parents in wheat breeding programs. Although the origins and pedigrees of Jimai 22 and Liangxing 99 are different, they are highly similar in many important agronomic traits, yield-associated traits, and so on. To identify the genomic differences between the two varieties, we performed whole-genome sequencing using the Illumina HiSeq2500 platform, with an average sequencing depth of 5.8×. We aligned the raw sequencing data against the Chinese Spring reference genome and identified the difference of copy-number variation (CNV) regions, single-nucleotide polymorphisms (SNPs) and InDels in sequence between the two varieties. Lengths of 466 Mb CNV intervals were shared by the two varieties. The lengths of cultivar-specific CNV intervals in Jimai 22 and Liangxing 99 were 91 Mb and 45 Mb, respectively, and these intervals are mainly located on chromosomes 2B and 4B. Beyond the CNV intervals, 1,547,371 SNPs and 137,817 InDels were different between the two cultivars. Based on the distribution of SNP densities in the intervals, we identified the polymorphic hotspot regions on chromosomes 1D, 2B, and 4B, making up 14.2% of the whole genome. The sequences of five previous cloned dwarf genes and spike length related genes were investigated, and two genes located in the polymorphic hotspot regions were detected with the frame shift variations. This study provides an important guidance for evaluating the genetic differences between two wheat varieties in the genomic level, and also identified both genetic similarity regions and polymorphic hotspot regions between Jimai 22 and Liangxing 99, which provided a valuable genetic information for future genetic improvement utilizing Jimai 22 and Liangxing 99 as parents.
wheat; Jimai 22; Liangxing 99; whole-genome resequencing; SNP; CNV
本研究由国家重点研发计划项目(2018YFD0100803), 国家自然科学基金项目(31701415)和中央高校基本科研业务费专项资助。
This study was supported by the National Key Research and Development Program of China (2018YFD0100803), the National Natural Science Foundation of China (31701415), and the Chinese Universities Scientific Fund.
郭伟龙, E-mail: guoweilong@cau.edu.cn; 宿振起, E-mail: suzhenqi80@163.com
E-mail: yangzhengzhao@cau.edu.cn
2020-01-15;
2020-06-02;
2020-08-10.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20200810.1525.002.html
10.3724/SP.J.1006.2020.01009
