新生儿Zellweger综合征1例的临床及基因诊断
2020-11-26冯雪李基伍丽
冯雪 李基 伍丽
(深圳市儿童医院新生儿科,广东深圳518038)
Zellweger综合征(Zellweger syndrome,ZWS)是一种罕见的常染色体隐性遗传性疾病,属于过氧化物酶体疾病。该病是一组过氧化物酶体功能受损的异质性遗传性代谢病,在大多数情况下,会导致不同程度的神经功能障碍[1]。过氧化物酶体疾病分为两个主要的类别[2]:过氧化物酶体生物发生异常及单一过氧化物酶体酶缺乏。ZWS属于前者,是婴儿早期最常见、最严重的过氧化物酶体疾病。发病率约为1/50,000[3]。新生儿主要表现为神经系统异常:肌张力低下、喂养困难、新生儿惊厥,头面部畸形如大囟门、前额突出、眼距过宽,肝脏异常如肝大、黄疸、胆汁淤积及肾囊肿等。ZWS临床表现复杂,缺乏特异性。本研究回顾性分析我院收治的1例ZWS新生儿临床资料并结合全外显子基因检测结果及相关文献进行复习,以探讨其临床及基因诊断、鉴别诊断,加深临床医生对本病的认识。
1 病例资料
1.1 入院情况:患儿女,因“肌张力低下19天”入院。系G2P2,胎龄37+5周,顺产娩出,出生体重2350g,Apgar评分1分钟8分,5分钟8分,10分钟9分。父母非近亲结婚。1兄体健。入院查体:体重2.25kg,反应可,哭声低。前囟4cm×5cm,张力正常。左侧眼裂较右侧小,口周无发绀,高腭弓,心肺查体无特殊。腹饱满,肝脾无肿大。双手垂腕,双足内翻,双下肢无浮肿,四肢肌力2~3级,肌张力减低。原始反射存在。
1.2 辅助检查:血、尿、便常规、心肌酶、肾功、电解质、凝血四项、TORCH、血、尿CMV-DNA、性激素五项、肾上腺皮质激素六项未见异常。肝功能:GGT 280IU/L、AST 532 IU/L、ALT 354 IU/L 7d 复查:总胆汁酸测定 111.0μmol/L、GTT 341IU/L、AST 178IU/L、ALT 147IU/L。胸片:双肺纹理增多。超声检查:心脏房水平左向右分流(2mm),左室整体舒张及收缩指标正常。消化道:考虑先天性胆管扩张症。盆腔:双侧卵巢未显示。泌尿系:考虑双肾多囊性病变。肌电图:各神经起初潜伏期延长,波幅降低、速度减慢。双侧正中、右侧尺神经波幅降低、速度减轻减慢或大致正常。选择性查双侧胫前肌EMG:未见异常。眼底检查未见异常。血、遗传代谢病筛查无明显异常。颅脑MR:双侧大脑半球可疑神经元移行障碍。吸吮吞咽评估功能不良。
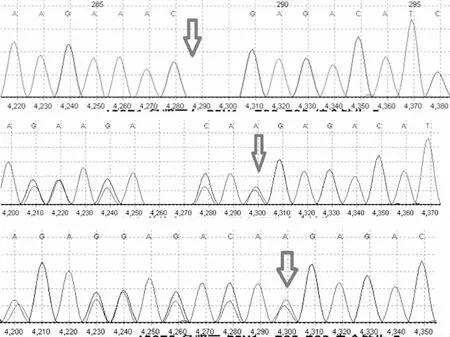
图1 患儿及其父母基因测序图(箭头所示为突变位点)
1.3 诊治经过:患儿生后即出现肌张力低下,吞咽及吸吮障碍,出生16d出现谷丙转氨酶升高。入院后予保暖、箱内吸氧、静脉营养、深度水解配方奶管饲,熊去氧胆酸利胆、吸吮吞咽康复训练等治疗,患儿仍肌张力低下、喂养困难,入院19d家长考虑预后不佳自动出院。随访至2月龄,婴儿出现口角抽动、眨眼及肢体抖动,家长未予治疗。
2 基因检测分析
PEX1基因(染色体位置 chr7:92147046,参考基因组hg19)第5个外显子发生纯合变异:(NM_000466.3):c.782-783delAA。该变异为移码突变,对应编码蛋白质自第261氨基酸Gln开始发生移码,并使得蛋白质翻译提前终止。预测可能导致所编码的蛋白质发生截短从而丧失其正常功能。家系验证患儿父母均为杂合携带,详见图1。该变异在参考人群数据库中频率极低,其中在ExAC外显子数据库中频率为0.00004175(其中东亚人群的频率为0.0000648),在gnomAD外显子数据库中频率为0.0000361(其中东亚人群的频率为0.0001089)。dbSNP154数据库有收录(rs749067142)。依据ACMG变异分类指南,这个变异为“2类-可能致病”。
3 讨论
过氧化物酶体病属遗传性极长链脂肪酸代谢病。过氧化物酶体可将极长链脂肪酸(Very long chain fatty acid,VLCFA)的 β-氧化,以及催化过氧化氢分解[4]。过氧化物酶体是VLCFA被氧化的唯一部位。过氧化物酶体疾病的主要神经病理学特征为神经元迁移和分化缺陷[5]。本例颅脑MR提示双侧大脑半球可疑神经元移行障碍,符合该病神经病理学特征。
过氧化物酶体疾病的临床表现随年龄而变化[6]:新生儿期特征性的表现为肌张力过低、癫痫发作、颅面畸形和骨骼异常。文献复习结果显示[7],新生儿惊厥发生率明显低于肌张力低下,肌张力低下可能是新生儿ZWS典型表现。本例患儿住院期间亦无明显惊厥。因此ZWS新生儿应与缺氧缺血性脊髓病、先天性感染、脊髓性肌萎缩(SMA)等肌张力低下的疾病鉴别。另外血浆VLCFA检测对于ZWS的诊断具有重要意义。
PEXl基因变异是ZWS起病的最主要原因。本例PEX1基因(染色体位置chr7:92147046,参考基因组hg19)第5个外显子发生纯合变异:(NM_000466.3):c.782-783delAA。该变异为移码突变,对应编码蛋白质自第261氨基酸Gln开始发生移码,并使得蛋白质翻译提前终止。文献复习结果显示新生儿期起病的ZSD患儿携带的变异类型大多为移码变异,该类变异对于蛋白功能影响较大,与ZSD的严重表型及不良预后密切相关。因此,遗传学检测及基因型表型分析对于ZSD的诊断以及预后判断具有重要意义[9]。
目前,Zellweger综合征暂无特效治疗方法。PEX基因检测是早期诊断、早期治疗以及产前咨询、产前诊断的关键,胃造瘘,提供热量,补充维生素,胆酸及应用抗癫痫药物,监测肝功能,凝血功能可延长ZWS患儿生存期。
