宏转录组测序揭示褐土脲酶基因的表达丰度和细菌来源
2020-11-25胡利伟轩贝贝戴华鑫郭建华牟文君蔡宪杰奚家勤薛超群宋纪真
胡利伟,轩贝贝,戴华鑫,郭建华,牟文君,刘 阳,翟 振,闫 鼎,蔡宪杰,奚家勤,薛超群,宋纪真*
1. 中国烟草总公司郑州烟草研究院,郑州高新技术产业开发区枫杨街2 号 450001
2. 上海烟草集团有限责任公司采购中心,上海市杨浦区长阳路717 号 200082
脲酶是氮素循环过程中的一种关键的含镍寡聚酶,土壤脲酶直接参与土壤中含氮有机化合物的转化,其活性可表征土壤中的氮素状况[1-2]。一般认为,土壤脲酶的酶活性与土壤中有机物的含量和土壤微生物的数量等正相关[3],多地实验表明施加有机肥比无机肥的烟田土壤脲酶酶活更高,施用有机肥使土壤脲酶活性稳定并维持较高水平[4-6]。土壤脲酶的活性随着烟草的生育期变化,不同时期变化速率不同,烟田土壤的脲酶活性从团棵期到旺长期缓慢增强,从旺长期到现蕾期增速加快,但从现蕾期到采烤前期快速下降,之后于采烤的后期上升至旺长期水平[7-8]。脲酶活性的变化反映了植烟土壤氮素的转化、积累、供应效应与土壤保肥性之间的协调性[9]。烟田土壤脲酶活性与烟株发病程度也有一定的相关性,例如随着黑胫病发病程度加重,烟株根际脲酶活性降低[10]。
土壤脲酶来自土壤微生物,如土壤细菌、真菌等。细菌在土壤中的种类和数量较多, 并具有较强的产酶能力[11-12],脲酶活性细菌是与土壤脲酶的性质和数量有直接关系的一类微生物[13-14]。近年来已经从土壤中分离出许多产脲酶菌。张知晓等[15]分离的土壤脲酶活性细菌分属于厚壁菌门(Firmicutes)和变形菌门(Proteobacteria)2 大类群,包括厚壁菌门的杆菌属(Bacillus)、芽孢八叠球菌属(Sporosarcina)和芽孢杆菌属(Lysinibacillus)以及变形菌门的产碱杆菌属(Alcaligenes)和假单胞菌属(Pseudomonas)等,认为土壤脲酶活性细菌优势类群是厚壁菌门,同时也存在变形菌门菌株。新西兰学者的研究表明枸橼酸杆菌等9 种细菌以及瓜枝孢菌等24 种真菌参与了牧场土壤中尿素的代谢[16]。也有学者从苗圃根际土壤分离到芽孢杆菌(Bacillus)和芽孢八叠球菌(Sporosarcina)等产脲酶菌株[17-18]。这些微生物编码的脲酶基因包括结构基因 ureA、ureB、ureC 和辅助基因 ureD、ureH、ureE、ureF 和 ureG 等,其中结构基因 ureA、ureB、ureC 分别编码γ、β、α亚基,组成尿素酶原,而辅助基因编码蛋白协助将Ni2+传递至脲酶活性中心,激活酶原[19]。
褐土在河南省境内主要分布于沙河与伏牛山一线之北,京广线以西的偏西北区域[20],自然状态下褐土的有机质含量多为4%~8%,耕种土壤在2%以下。之前的宏基因组测序结果显示河南地区褐土中产脲酶微生物主要为链霉菌、节杆菌和不动杆菌等细菌,足马杜拉分枝菌等真菌以及亚硝化球菌等古细菌[21]。这些微生物编码的脲酶基因种类繁多,但是其中哪些脲酶基因表达能最终发挥作用尚不清楚。 宏转录组测序技术(Metatranscriptomic sequencing)通过对特定时期、特定环境样品中所有微生物群落的转录本进行大规模高通量测序,可以直接获得环境中可培养和不可培养的微生物基因表达信息,目前已经越来越多地在环境微生物学,医学微生物等领域应用[22-24]。为此,对来自河南省平顶山、洛阳、许昌、郑州和安阳等市的5 个高脲酶活性土壤样本富集培养,进行土壤微生物RNA 提取、建库及转录组测序,分析脲酶基因在转录水平的表达丰度,并对微生物来源进行解析,旨在明确褐土尿素代谢中微生物的作用。
1 材料与方法
1.1 土壤样品的采集
采样时间为2018 年6~10 月,采样地点为郑州市上街区和荥阳市、洛阳市洛宁县、平顶山市郏县、许昌市襄城县及安阳市林州市的农田。取样深度为0~10 cm 土壤,采用5 点混合法进行土壤取样,剔除杂物后装入塑料自封袋,置于10~15 ℃通风条件良好处保存。每个地级市取样10份,共取土样50 份,对其脲酶活性进行检测[21],选取各市脲酶活性最高的土壤样品进行下一步实验。
1.2 产脲酶菌的富集培养
洛阳、许昌、郑州、安阳和平顶山脲酶活性最高的土壤样品分别为 XC-10、ZZ-07、AY-06、LY-07和PDS-01,每个样品取样3 次(10 g/份),共15 份,分别加入含有50 mL液体培养基(蛋白胨1.0 g/L、氯化钠1.2 g/L、磷酸二氢钾 0.8 g/L、葡萄糖 1.5 g/L、硫酸镍 0.1 g/L、酚红0.016 g/L、尿素20 g/L,pH 7.0)[25]的250 mL 三角瓶中,在30 ℃摇床中180 r/min振荡培养24 h,接着将15 瓶富集培养产物进行混合,用液氮冰冻后放入-70 ℃冰箱保存。
1.3 土壤RNA 的提取
使用Powersoil 土壤总RNA 提取试剂盒(美国Mobio 公司)进行土壤RNA 的提取,操作按照试剂盒说明书进行。采用琼脂糖凝胶电泳检测RNA的完整度并使用紫外分光光度计(美国Nanodrop公司)检测RNA 的纯度和浓度,将提取的RNA 置于-70 ℃冰箱中保存。
1.4 RNA 的逆转录与建库
利用DNA 探针与RNaseH 酶促反应特点,采用Ribo-off rRNA 去除试剂盒(南京诺唯赞生物科技有限公司)对土壤微生物总RNA 进行处理,以获得能够满足后续文库构建的mRNA 及其他非编码RNA 产物,接着采用VAHTS™Stranded mRNA-seq Library Prep Kit for Illumina®试剂盒(南京诺唯赞生物科技有限公司)对产物进行逆转录、双链cDNA 合成、cDNA 片段化修饰、磁珠纯化及片段化分选、文库扩增等处理。文库用8%的PAGE 胶电泳检测,利用Qubit 2.0 DNA 检测试剂盒对回收的DNA 进行精准定量。
1.5 测序及生物信息学分析
使用Hiseq 测序仪(美国Illumina 公司)进行双端测序,双向各测150 bp。对原始数据质量值等信息进行统计,并使用FastQC 软件对样本的测序数据质量进行可视化评估。使用Trimmomatic 软件[26]去除带 N 碱基的序列、reads 中的接头序列和低质碱基,随机从Clean 数据中抽取10 000 条序列与 NCBI NT 数据库进行 BLASTN 比对,取e-value ≤1e -10 并且相似度>90%、覆盖度>80%的比对结果,计算其物种分布,进行污染检测。使用Trinity软件[27]将clean数据de novo组装成转录本,参数min_kmer_cov 2,其余默认。使用TransDecoder软件预测Unigene 的CDS 并翻译成蛋白序列。将获得的基因和蛋白序列与包括NR、COG、eggNOG等在内的相关数据库进行比对,获得基因的功能及物种注释信息[28-30]。将拼接得到的转录本作为参考序列,使用Bowtie2 将质控后的测序序列与参考序列进行比对,并通过RSeQC 软件统计比对结果[31]。计算每个特异基因的TPM(每百万序列转录本数,Transcripts per million)值以分析其表达丰度。
2 实验结果
2.1 褐土产脲酶微生物的宏转录组测序
对各地区脲酶活性最高的土壤样品XC10、ZZ07、AY06、LY07 和PDS01 进行富集培养,提取土壤RNA 后进行宏转录组测序和分析,初步获得总数据量14 187 012 298 bp,拼接后获得数据量271 Mb 及1 106 689 个转录本,预测特异基因940 402个,N50 为 262 bp,最大基因长度 18 629 bp,平均长度288.18 bp(表1)。

表1 转录组测序的相关结果Tab.1 Results of metatranscriptomic sequencing
2.2 转录本的微生物来源
富集后转录本与NCBI 数据库进行比对,结果表明序列匹配数超过10 条的有4 567 种菌株,序列匹配数在10~99 条之间的菌株种数最多,为3 636种,序列匹配数在200~299 条之间的有154 种,序列匹配数在300~399 条之间的有92 种,序列匹配数量随着菌株种数的增多基本呈现减少的趋势,最多的匹配数为11 408 条序列(图1),其中丰度最高的10 个菌株为Acidobacteria bacterium DSM 100886、 Brevibacillus choshinensis、 Agromyces sp.NDB4Y10、 Candidatus Rokubacteria bacterium CSP1-6、Bacillus fordii、Gemmatirosa kalamazoonesis、Comamonas testosteroni、Arthrobacter sp. Soil736、Cupriavidus necator 和Sporosarcina koreensis 等(图 2)。
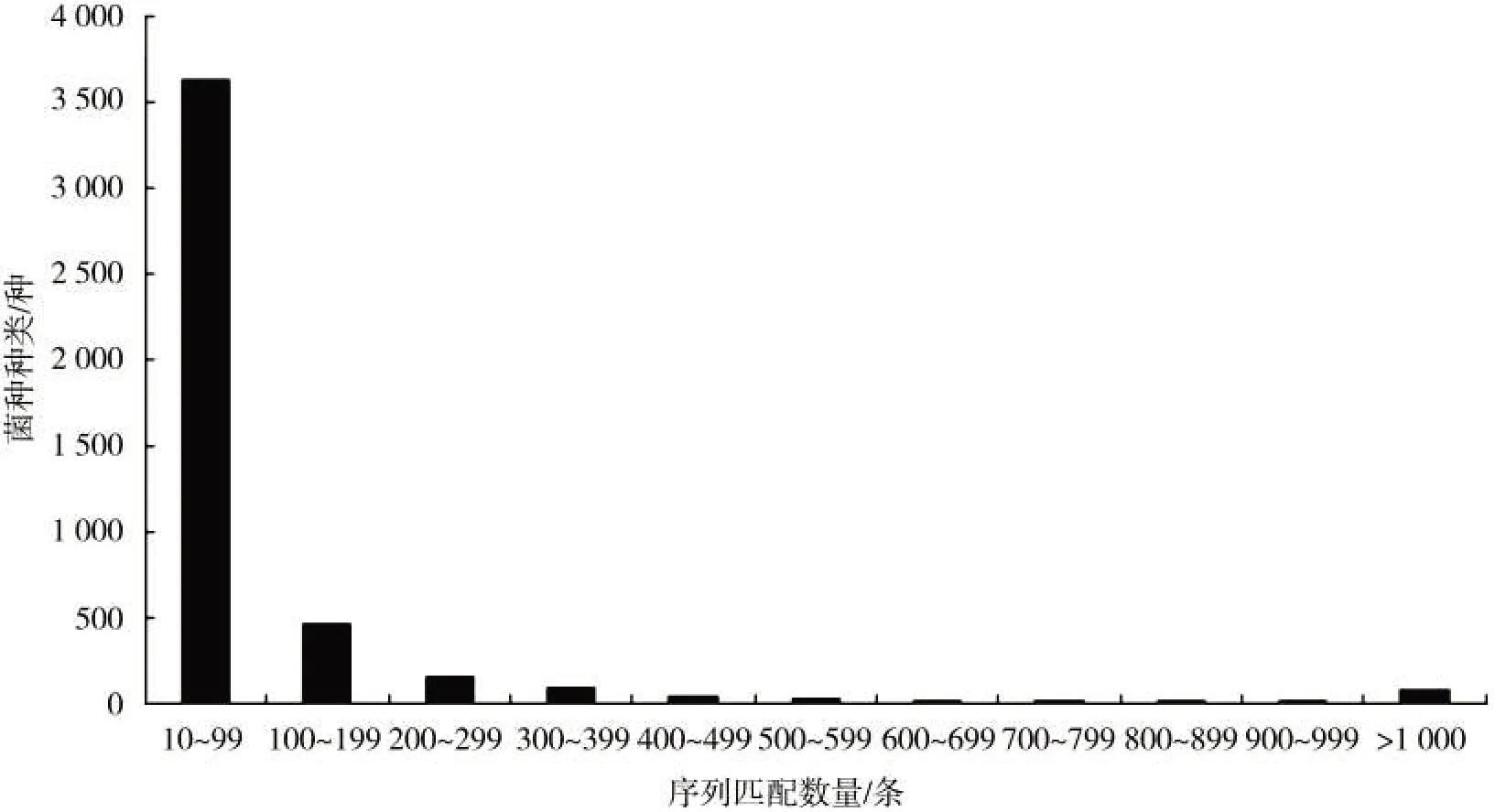
图1 序列匹配的菌株数量分布Fig.1 Quantity distribution of bacteria with matched sequences
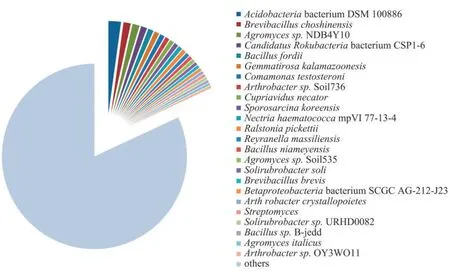
图2 土壤RNA 转录本的微生物来源Fig.2 Microbe sources of soil RNA transcripts
2.3 脲酶基因的筛选与注释
通过注释筛选3 个及以上拷贝的转录本,共获得122 个脲酶结构基因与115 个脲酶辅助基因。其中脲酶结构基因包含93 个ureC(α亚基)、16 个ureB(β亚基)及13 个ureA(γ亚基);脲酶辅助基因中有 29 个 ureD、12 个 ureE、36 个 ureF、29 个 ureG、5个ureH 和4 个ureJ。利用脲酶结构基因与辅助基因编码的蛋白序列对NCBI 的非冗余蛋白库(Non-redundant database,NR)进行 BLASTP 分析,结果(图3)表明,122 个脲酶结构蛋白和NCBI 的NR 库收录序列的同源性为78%~100%,其中脲酶结构蛋白α、β和γ亚基与NR 库收录序列的同源性在90%~100%之间的分别有75、5 和12 个。115个脲酶辅助蛋白和NR 库收录序列的同源性为64.25%~99.95%,UreD、UreE、UreF、UreG、UreH及UreJ 和NR 库收录序列的同源性在90%~100%之间的分别有7、5、9、20、1 和3 个。而UreD、UreF和NR 库收录序列相似度低于80%的蛋白较多,分别有 11 个和 13 个。
2.4 脲酶基因的表达情况

图3 脲酶亚基和NR 数据库比对的同源性分布Fig.3 Distribution of homology of urease subunits and those in NR database
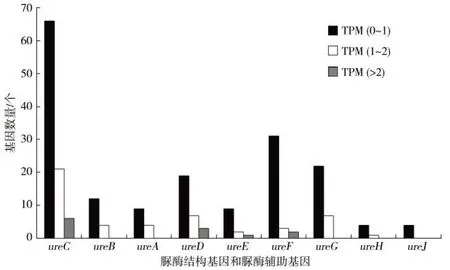
图4 脲酶基因的表达情况Fig.4 Expression levels of urease genes
如图4 所示,ureC、ureB、ureA、ureD、ureE、ureF、ureG、ureH 和ureJ 等脲酶基因中,编码α亚基的ureC基因数量最多,表达值TPM 在0~1 之间的有66个,1~2 之间的有 21 个,大于 2 的有 6 个,最高的DN897996_c0_g1 为 3.16。编码β亚基的 ureB 基因有 16 个,表达值 TPM 在 0~1 之间的有 12 个,1~2之间的有4 个。编码γ亚基的 ureA 基因有13 个,表达值 TPM 在 0~1 之间的有 9 个,1~2 之间的有 4个。另外ureD、ureE 和ureF 的表达值最高的分别为 5.31、2.00 和 2.71,其中 ureD 表达值 TPM 超过 2的有 3 个转录本,分别为 DN911511_c0_g1(2.03)、DN893873_c0_g1(3.31)和 DN907166_c3_g2(5.31)。ureH 和ureJ 数量较少,表达值也较低。
2.5 脲酶基因的微生物来源
脲酶基因的属来源如表2 所示。芽孢杆菌属Bacillus、芽孢八叠球菌属Sporosarcina、节杆菌属Arthrobacter、链霉属Streptomyces 和类芽孢杆菌属Paenibacillus 等属细菌具有较多的脲酶结构基因和辅助基因在转录水平表达,芽孢杆菌属Bacillus 中有39 个脲酶基因在转录水平表达,其中结构基因ureC、ureB 和 ureA 各有 8、3 和 1 个表达,ureD、ureE、ureF、ureG 和ureH 等脲酶辅助基因分别有10、3、6、6 和 2 个表达在mRNA 水平被检测到。芽孢八叠球菌属Sporosarcina 中有24 个脲酶结构基因和辅助基因在 RNA 水平表达,包括 ureC、ureB、ureD、ureE、ureF 和 ureG 等,其中脲酶辅助基因 ureC 和ureF 数量最多,分别为9 个和7 个。节杆菌属Arthrobacter 中 ureC、ureB、ureD、ureF 和 ureG 等脲酶基因,分别有 5、2、4、6 和 3 个基因在转录水平表达。 链霉属Streptomyces细菌在RNA水平表达的有ureC、ureB、ureA、ureD、ureF 和ureG 等基因,其中ureC较多,有10 个基因。此外,还发现了Paenibacillus、Candidatus、 Bradyrhizobium、 Pseudarthrobacter、Ralstonia 及Cupriavidus 等属细菌的脲酶结构基因和辅助基因转录的mRNA。
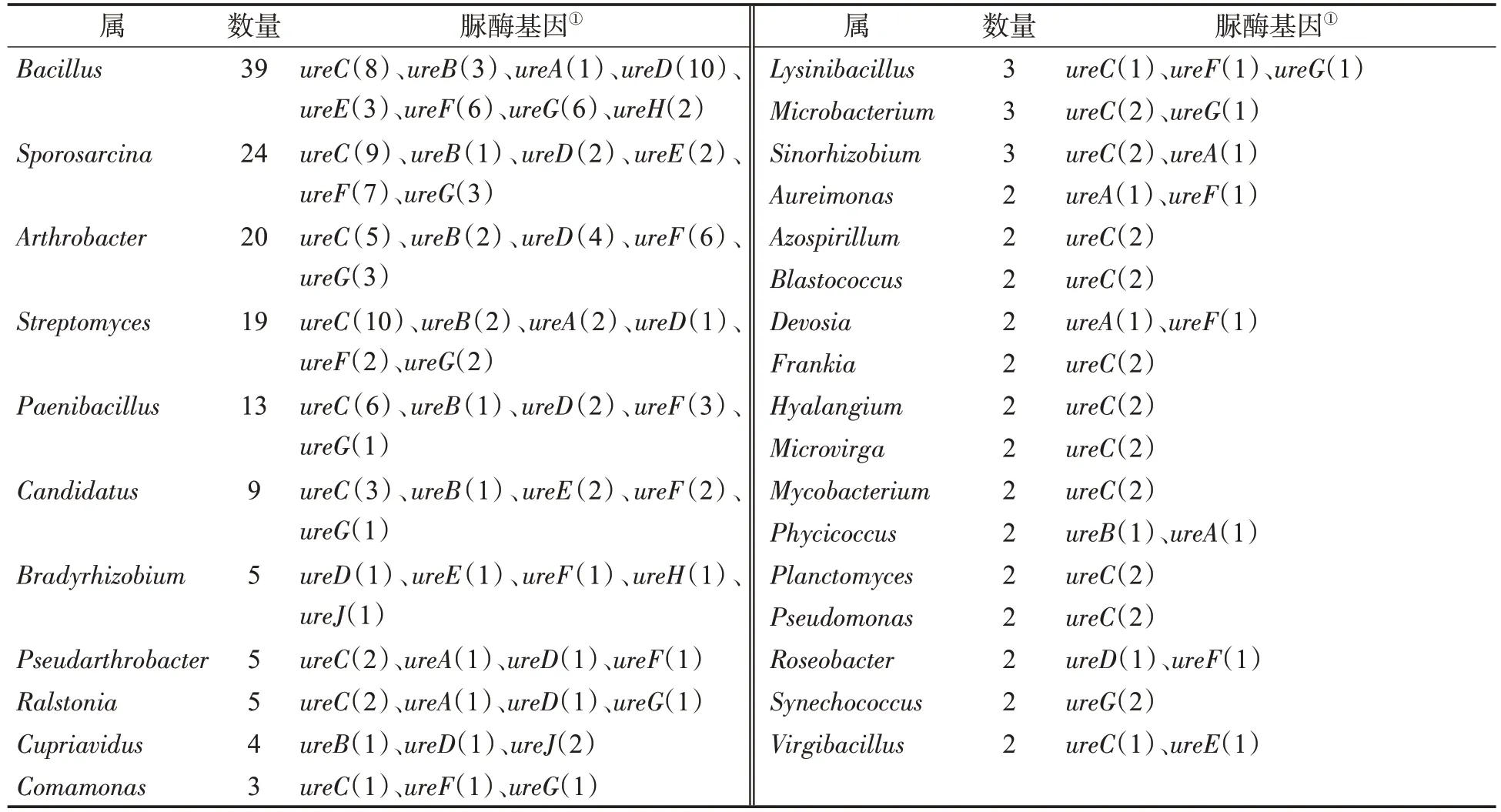
表2 细菌脲酶基因的属来源Tab.2 Genus sources of urease genes in bacteria (个)
3 讨论
张知晓等[15]研究发现从云南昆明市农田土壤分离出的9 种产脲酶菌分别来自厚壁菌门的杆菌属、芽孢八叠菌属、芽孢杆菌属以及变形菌门的产碱杆菌属、假单胞菌属等。沈琼雯[32]从贵州喀斯特地区的160 份土壤中成功鉴定了343 株产脲酶细菌,有25 属79 种,大部分为革兰氏阳性菌,其中优势菌属是芽孢杆菌属、肠产气杆菌属、假单孢菌属和类芽孢杆菌属,分别占比15.19%、12.66%、12.66%和5.06%。之前的研究采用宏基因组测序方法对河南省代表性褐土中的脲酶微生物来源进行了分析,发现褐土脲酶中主要细菌来源有芽孢杆菌属、节杆菌属、链霉菌属、芽孢八叠球菌属、不动杆菌属、类芽孢杆菌属和假单胞菌属等,产脲酶真菌与足马杜拉分枝菌、蓝莓枝枯病菌和嗜热毛壳菌等亲缘关系较近,古细菌中亚硝化球菌、氨氧化古菌和奇古菌等可能也参与了褐土中尿素的降解[21]。本研究通过 BLAST 比对分析,在 mRNA 水平表达的脲酶结构基因和辅助基因与芽孢杆菌属Bacillus、芽孢八叠球菌属Sporosarcina、节杆菌属Arthrobacter、链霉属 Streptomyces、类芽孢杆菌属Paenibacillus、念珠菌属Candidatus、类芽孢杆菌属Bacillales、慢生根瘤菌Bradyrhizobium、假节杆菌属Pseudarthrobacter、劳尔氏菌 Ralstonia、贪铜菌属Cupriavidus、丛毛单胞菌属Comamonas、赖氨酸芽孢杆菌属Lysinibacillus、细杆菌属Microbacterium 和中华根瘤菌属Sinorhizobium 等细菌的亲缘关系最近。未检测到编码脲酶的真菌和古细菌的存在,可能是因为细菌在褐土氮素供应状态转化中作用更 为 关 键 。 Bacillus、Sporosarcina、Arthrobacter、Streptomyces 和 Paenibacillus 等 5 个属 mRNA 水平表达的脲酶结构基因和辅助基因总共108 个,占所有表达脲酶基因的45.6%,这些属的细菌在褐土脲酶活性的发挥中起着重要的作用,下一步将对它们的特异抑制剂进行重点研究。
细菌脲酶是由2~3 个不同亚基组成的杂聚肽,其中α亚基在其中起到至关重要的作用。褐土的宏转录组测序分析显示编码脲酶结构蛋白α亚基的ureC 基因数量最多,为93 个,这些基因在RNA 水平也维持较高的表达丰度,说明ureC 在褐土中尿素的代谢中有着较为重要的作用,其高表达也与褐土的高脲酶活性一致。UreF 和UreG 形成一个复合物,使酶原对镍离子处于感受态,促进镍离子有效地结合到活性位点。有研究表明ureF基因的敲除能够降低脲酶活性[33],这里褐土中ureF 基因表达丰度较高,其丰度最高的转录本TPM 值达到了5.37,ureF 的高表达可能是褐土脲酶活性较高的原因之一。UreD 在脲酶金属中心组装过程中是镍插入位点的促进剂也是招募其他辅助蛋白的支点,有研究表明产气克雷伯氏菌的UreD 是可溶的,并结合2 个当量的镍离子,UreD不存在时无法激活脲酶[2]。褐土微生物中ureD 基因的数量和表达值都相对较高,可能与其在脲酶活性发挥中的重要角色有关。而其他基因ureB、ureA、ureE、ureH 及 ureJ 数量相对较少,说明其在脲酶复合体的组成中占比可能较低,在脲酶酶活性中的作用相对较小。
4 结论
通过褐土微生物RNA 的宏转录组测序,共筛选到转录水平表达的122 个脲酶结构基因(93 个ureC、16 个 ureB 和 13 个 ureA)以及 115 个脲酶辅助基因(29 个 ureD、12 个 ureE、36 个ureF、29 个 ureG、5 个 ureH 和 4 个 ureJ)。ureB、ureA、ureG、ureH 和ureJ 等基因在RNA 水平的表达相对较低,TPM 值都在 2 以下,ureC、ureD、ureE 和 ureF 等基因的 TPM值绝大多数都在2 以下,个别高于2。细菌来源的芽孢杆菌属Bacillus,芽孢八叠球菌属Sporosarcina、节杆菌属Arthrobacter、链霉属Streptomyces 和类芽孢杆菌属Paenibacillus 等5 个属在褐土脲酶酶活的发挥中贡献度较大。
