化学链过程中Cu低浓度掺杂改性Fe-基载氧体反应性能:实验与理论模拟
2020-11-18袁妮妮白红存安梅胡修德郭庆杰
袁妮妮,白红存,安梅,胡修德,郭庆杰
(宁夏大学省部共建煤炭高效利用与绿色化工国家重点实验室,化学化工学院,宁夏银川750021)
引 言
高效载氧体是当前化学链技术从基础研究实现工业应用的重要物质基础。化学链技术[1]是一种新型能源高效、清洁转化利用技术,具有CO2有效捕集优势。该技术借助载氧体[2-3]氧传输和载热作用,分别与燃料和空气在燃料反应器和空气反应器中反应,中间省去空分装置,实现燃料清洁、高效转化,可有效控制污染物的产生和排放。因此,化学链技术具有广阔的应用前景[4-5]。实现该技术的关键是高效载氧体的选择。高效载氧体要求载氧率高、反应性能好和循环性能稳定。因此,高效载氧体可减少化学链过程中载氧体用量,降低设备负荷,缩短燃料在反应器内转化的时间。
针对载氧体进行设计制备,实现性能提升仍是目前化学链领域的重要研究课题。当前在化学链技术规模化利用中,研究最广泛的为Fe-基载氧体材料[3,6-7]。其优点主要表现为价格低廉、储量丰富、环境友好、无毒性,且热力学稳定性高、力学性能好等。因此,Fe-基载氧体材料有望成为具有工业应用前景的优选材料。但是,Fe-基载氧体反应活性相对较低,限制其广泛应用。至今多个研究团队对Fe-基载氧体进行了设计制备和化学链过程中反应性能的研究。例如王保文等[8-9]采用溶胶-凝胶燃烧法制备Fe2O3/Al2O3载氧体,通过加入惰性组分Al2O3增大载氧体表面积,优化孔径分布,改善载氧体颗粒微观结构。程煜等[10]采用浸渍法制备Fe4Al6、Fe4Al6Ni1、Fe4Al6K1 等 一 系 列Al、Ni、K 等 改 性Fe-基载氧体,对Fe-基载氧体组成进行了改性,对煤化学链气化过程具有促进作用。杨明明等[11]通过溶胶-凝胶法制备ATP 负载Fe-基载氧体,改善载氧体孔隙结构和表面积,使载氧体反应活性增强,提高了煤转化率。Bhavsar 等[12]制备Fe@SiO2核壳结构载氧体,在化学链应用过程中,由于Fe2SiO4形成以及核壳结构破坏,使载氧体性能降低。虽然对Fe-基载氧体从制备方法、组成、结构等方面的研究较多,但获得的单组分Fe-基载氧体在使用过程中仍面临反应活性低、性能衰减快、循环稳定性差等问题。
目前报道较多的单金属载氧体主要包括Fe2O3、CuO、NiO、Mn2O3等,其各具优缺点。因此,可考虑将不同金属与Fe-基载氧体组合,得到复合Fe-基载氧体。利用多元金属之间互补和协同作用,设计Ni-Fe[13]、Cu-Fe[14]、Ca-Fe[15]、Mn-Fe[16]等复合载氧体,可提高Fe-基载氧体综合性能。例如吴宪爽等[13]制备Ni-Fe 和NiFe2O4复合载氧体,由于NiO 反应活性高,可以弥补Fe-基载氧体反应活性低的不足,2种载氧体用于化学链制氢中表现出良好性能,但循环过程中出现烧结。Jiang等[17]研究CuO添加量对Cu-Fe载氧体结构和性能影响,研究表明CuO 加入使Fe-基载氧体成多孔结构,增强载氧体反应活性,但20%CuO 加入时,载氧体出现烧结。Niu 等[18]研究发现CuO 和赤铁矿混合形成的CuFe2O4载氧体具有金属协同效应,可提高赤铁矿反应活性,并使CuO 在化学链过程中稳定性增加。Frick 等[19]对Cu-Fe 载氧体颗粒进行物理化学表征研究,表明该载氧体在足够强度下,与CO 和CH4具有较高的反应活性,适用于化学链燃烧过程。Qin 等[20]报道化学链气化过程中1%(mol)Cu 掺杂改性Fe-基载氧体,在700℃下,CH4转化制合成气,载氧体转化率提高了470 %,通过计算,与1000℃下相比,节约能量约35 %。因此,选择Cu 作为掺杂剂,低浓度掺杂改性Fe-基载氧体具有一定优势。然而,关于Cu 低浓度掺杂改性Fe-基载氧体相关研究仍然有待进一步深入,尤其缺乏从分子或原子水平上对Cu 低浓度掺杂改性Fe-基载氧体的反应性能和微观机理的系统研究。
本文借助TGA 实验和DFT 计算,对Fe2O3载氧体晶格内Cu 低浓度[1%(mol)]掺杂改性Fe2O3载氧体(Cu-Fe2O3)反应性能和微观分子反应机理进行系统研究。结合XRD和SEM-EDX 等对Cu-Fe2O3载氧体组成、结构和表面元素分布进行物性表征,从微观角度探究Cu-Fe2O3载氧体反应机理。期望本研究对Fe-基载氧体筛选、设计和优化具有理论指导意义[21]。
1 实验与理论计算方法
1.1 载氧体制备
本文采用共沉淀法制备Cu-Fe2O3载氧体,所用试剂:Fe(NO3)3·9H2O 和Cu(NO3)2·3H2O,分别将0.1 mol Cu(NO3)2·3H2O 和9.9 mol Fe(NO3)3·9H2O 溶 于500 ml去离子水中,搅拌溶解均匀,为溶液A。并配制1 mol/L Na2CO3共沉淀剂,为溶液B。通过蠕动泵将溶液A、B 均匀混合,并保持溶液pH 为9~10。待溶液沉淀完全后继续搅拌30 min,静置分层,并弃去上清液,用去离子水反复洗涤至中性,减压过滤,烘箱中120℃下恒温12 h,转入马弗炉900℃煅烧3 h,冷却并研磨筛分成75~150 μm 粒径颗粒,样品密封备用。按照上述方法制备Fe2O3空白样品。
1.2 载氧体性能测试
采用德国耐驰STA 449F3 常压热重分析仪(thermogravimetric analyzer,TGA),以10℃/min 升温速率分别从室温升至700、750、800、850、900℃终温,恒温50 min,恒温阶段通入200 ml/min 还原性气体(20%(vol) H2,Ar 平衡),保护气为Ar,流速为20 ml/min。
1.3 样品表征
采用X 射线衍射仪(XRD,Bruker :D8 Advanc A25)分析载氧体组成和晶体结构,操作条件为:Cu Kα辐射(λ=0.154 nm),工作电流40 mA,工作电压40 kV,衍射角2θ 范围为5°~85°,扫描速度为2(°)/min。
采用电子扫描显微镜(SEM,德国卡尔蔡司ZEISS EVO18)和能量色散X射线光谱(EDX)技术,对载氧体微观形貌和表面元素分布进行表征。扫描加速电压10 kV,分辨率为1 μm。
1.4 密度泛函理论计算
基于密度泛函理论(DFT)计算方法,在ADF(Amsterdam density functional) 计算平台中BAND 周期性模块完成计算[21-23]。与文献[20,24-27]中采用的GGA-PBE 方法相比,ADF 中集成的GGA 方法,使用Slater基函数对原子轨道的描述更接近真实体系,且计算精度高、效率快,对处理周期性过渡金属体系具有明显优势。采用DZP 基组进行结构优化和反应机理研究,DZP 基组用一个极化函数对双Zeta 基组函数扩展得到,可对本文计算模型进行较好处理。采用NEB(nudged elastic band)方法对H2在载氧体表面反应机理进行研究,文献[20,28-29]中多次报道,采用该方法对Slab 周期性平板模型表面反应机理进行研究。计算前,对Cu-Fe2O3载氧体表面H2反应机理研究中,能垒计算进行自旋极化测试,测试结果为:未考虑自旋极化时能垒为1.68 eV,考虑自旋极化后能垒为1.69 eV,相对偏差为0.77,偏差较小,综合考虑计算效率和精度,机理研究中反应能垒计算忽略自旋极化。自洽收敛标准为:(1)原子最大受力标准:0.01 Ha/Å;(2)能量收敛标准:0.001 Ha;(3)最大位移收敛标准:0.01 Bohr;(4)K-space 设置为:Normal。
选 取α -Fe2O3(001) 低 指 数 晶 面[30-32],并 以Fe—O3—Fe…为反应表面的稳定结构,以此为原胞,2×2 扩胞得到超胞模型。计算中设定底部5 层原子固定,表面4层原子和吸附分子弛豫,真空层厚度为20 Å(1 Å=0.1 nm),消除相邻平板层之间相互作用,并保证吸附分子有足够弛豫空间,得到Fe2O3周期性Slab 模型。该模型表面单个Fe 原子被Cu 原子取代,得到Cu 原子掺杂取代Cu-Fe2O3载氧体模型,如图1 所示。模型中晶胞参数(a=b=5.035 Å,c=13.720 Å)与实验样品晶胞参数(a=b=5.035 Å, c=13.747 Å)及文献[22]报道的吻合。
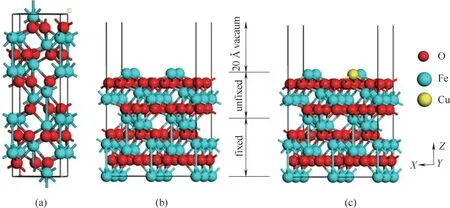
图1 α-Fe2O3原胞(a),α-Fe2O3(001)表面(b)和Cu-Fe2O3载氧体模型(c)Fig.1 Primitive cell of α-Fe2O3(a),α-Fe2O3(001)surface(b)and model of Cu-Fe2O3oxygen carrier(c)
2 结果与讨论
2.1 载氧体微观结构分析
通过XRD 和SEM-EDX 技术对载氧体组成、晶体结构和元素分布进行表征,如图2所示。由图2(a)可知,新鲜Fe2O3和Cu-Fe2O3载氧体均表现为Fe2O3的特征衍射峰,这表明载氧体经过高温煅烧后完全转化为Fe2O3相,未出现其他杂质相。此外,Cu-Fe2O3载氧体中掺杂的低浓度Cu原子完全进入Fe2O3晶格中替代等同数量的Fe原子[20]。因此,以Fe2O3为原胞构建本文计算模型,如图1(a)所示。Fe2O3载氧体晶格氧完全释放后转化为金属单质Fe,而Cu-Fe2O3载氧体晶格氧释放后转化为单质Fe 和单质Cu。图2(b)中,采用小角XRD 对Cu-Fe2O3载氧体晶胞结构进行分析。从图可知,Cu-Fe2O3载氧体衍射峰发生轻微红移,这是由于Cu 掺杂导致Fe2O3晶胞结构发生收缩所致。结果表明,晶胞参数a/b 从5.036 Å 收缩到5.035 Å,即Cu 掺杂较小幅度改变了Fe2O3载氧体晶胞大小,与文献[20]报道的吻合。
采用SEM-EDX Mapping对新鲜Cu-Fe2O3载氧体表面元素分布进行表征,如图2(c)所示。从图中可知,Cu-Fe2O3载氧体表面Cu、Fe、O元素分布均匀,未出现局部聚集或团聚现象。以上结果表明,采用共沉淀法实现Fe2O3晶格内原子尺度Cu低浓度[1%(mol)]掺杂取代,载氧体表面各元素分布均匀,并保持Fe2O3原有晶相结构。因而不易出现团聚和烧结现象。
2.2 载氧体反应性能分析
通过载氧体在还原性气氛晶格氧释放过程中转化率和晶格氧释放速率对其性能进行评价[27]。载氧体转化率Ct与失重量转化关系如式(1):


图2 载氧体颗粒表征:Fe2O3和Cu-Fe2O3载氧体与H2反应前后XRD谱图(a);Fe2O3和Cu-Fe2O3新鲜载氧体样品小角XRD谱图(b);Cu-Fe2O3新鲜载氧体样品SEM-EDX Mapping表征(c)Fig.2 Characterization of oxygen carrier particles:XRD patterns of Fe2O3 and Cu-Fe2O3 oxygen carrier before and after the reaction(a);small range XRD pattern of fresh Fe2O3 and Cu-Fe2O3 oxygen carrier(b);SEM-EDX Mapping images of Cu-Fe2O3 oxygen carrier(c)
式中,wi为起始样品质量;wt为t 时刻样品质量;wx为载氧体晶格氧完全释放后质量。从式(1)可知,单位时间载氧体转化率越高,晶格氧释放速率越快,反应时间越短,载氧体反应活性越强。
由式(1)可得载氧体在不同温度下转化率曲线,如图3 所示。由图3 可知,载氧体与H2反应时,随温度升高载氧体转化率逐渐增大,并且随温度升高反应起始阶段载氧体晶格氧释放速率逐渐增大,但随着反应进行晶格氧释放速率逐渐减小,Cu-Fe2O3载氧体转化率和晶格氧释放速率明显高于Fe2O3载氧体。900℃时,Cu-Fe2O3载氧体晶格氧完全释放还原为金属单质Cu 和Fe 时,比Fe2O3载氧体反应结束时间提前20 min 左右(两次实验条件基本相同),Cu-Fe2O3载氧体晶格氧释放时间明显缩短。以上结果表明:Cu 元素引入后Fe2O3载氧体转化率和晶格氧释放速率明显得到改善。
如图3 所示,根据不同温度下载氧体转化率曲线,由Arrhenius方程得到载氧体与H2反应的表观活化能Ea[26]。由图3 中活化能曲线可知,Cu 掺杂使Fe2O3载氧体Ea由83.9 kJ/mol 降低至72.3 kJ/mol,可见,Cu 掺杂使Fe2O3载氧体活化能明显降低,反应活性增强。因此,Cu 掺杂显著改善了Fe2O3载氧体反应活性。
综上,Cu 掺杂使Fe2O3载氧体转化率和晶格氧释放速率明显增大,Cu-Fe2O3载氧体反应性能显著优于Fe2O3载氧体。
2.3 密度泛函理论(DFT)计算
采用DFT 方法对Cu-Fe2O3载氧体微观结构和反应机理进行研究。Cu 掺杂使Fe2O3载氧体结构中键型和键长发生变化,表现为表面部分Fe-O 键(1.741 Å)和Fe-Fe 键(3.150 Å)分 别 被Cu-O 键(1.882 Å)和Cu-Fe 键(3.190 Å)取代,键长明显增大,表明Fe2O3载氧体微观结构发生改变。
基于DFT 方法对载氧体表面H2分子吸附性进行研究,主要考察不同相位吸附能和吸附距离。通过DFT计算H2在载氧体表面吸附能ΔEads:
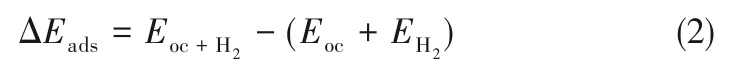
在吸附性研究中,H2分子在载氧体表面金属原子top、bridge、hollow 三种不同相位吸附性均予以考虑。按照本文1.4 节中计算条件,优化得到不同吸附相位稳定结构。由式(2)计算体系吸附能ΔEads,以对H2在载氧体表面稳定吸附相位和载氧体表面活性位点进行研究。
计算H2分子在Fe2O3和Cu-Fe2O3载氧体表面金属原子(Fe/Cu)不同吸附位吸附能。结果表明:H2在Fe-基载氧体表面均可发生稳定吸附,且不同位吸附距离不同。H2分子在Fe2O3载氧体表面Fe 原子top位、Cu-Fe2O3载氧体表面Cu原子top位和Fe原子top 位吸附能依次分别为-0.13、-0.03、-0.11 eV,均为物理吸附,相比bridge 和hollow 位,金属原子top位吸附作用最强,对应吸附距离依次分别为2.15、2.14、2.18 Å,吸附距离最远。因此,不同吸附相位中,载氧体表面金属原子top 位是H2分子吸附稳定相位,吸附也更容易发生。即H2分子垂直吸附于载氧体表面金属原子(Fe/Cu)top 位构象最稳定,也是H2分子在载氧体表面吸附主要形式,表明载氧体表面金属原子是其表面活性位点。
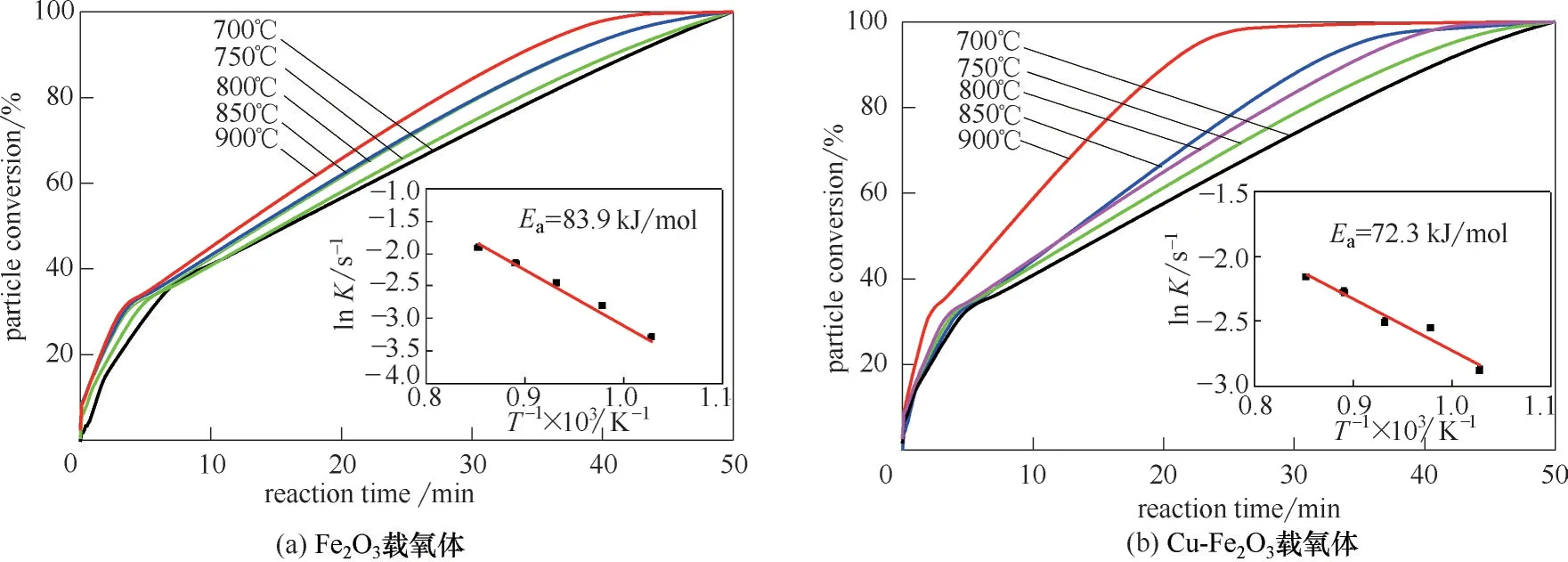
图3 载氧体与H2反应转化率及活化能曲线(插图)Fig.3 Conversion and Ea curves(inset)of oxygen carrier reaction with H2
H2在载氧体表面反应机理计算中,首先确定反应的起点和终点。这里以H2分子在载氧体表面稳定物理吸附为反应起点。该构型中H—H 键未断开,体系能量最低,即反应物(R)。反应完成后,载氧体表面吸附的H2分子中H—H 键断开,并与载氧体表面O 原子反应生成H2O,从载氧体表面脱附。该构型为反应终点,即产物(P)。按照本文1.4 节中计算条件,采用NEB 方法搜索过渡态结构(transition states,TS)。在反应机理计算时,为进一步对反应过程进行研究,计算反应能垒ΔEbarrier:

式中,ETS为反应过程中过渡态结构能量;ER为载氧体表面吸附H2分子后总能量。由式(3)可知,反应能垒ΔEbarrier是反应发生所需最低能量,ΔEbarrier越低,反应越容易发生。
H2与Cu-Fe2O3载氧体反应,考虑载氧体表面不同金属原子(Cu/Fe)位反应,如图4所示。由图4(a)可知,H2在Fe2O3载氧体表面反应包含Fe 原子top、bridge、hollow 位反应,反应能垒ΔEbarrier分别为2.30、2.47 和2.64 eV,top 位反应能垒最低,反应更容易发生,是其最优反应路径,这与载氧体表面吸附性结果相符。因此,Fe2O3载氧体表面Fe 原子是H2分子稳定吸附和能垒最低反应位点。类似地,如图4(b)、(c)所示,H2在Cu-Fe2O3载氧体表面Fe 和Cu 原子不同相位反应路径中,Cu 和Fe 原子top 位能垒均最低,分别为1.68 eV 和1.81 eV,也是top 位反应路径最优。三种反应中最优反应路径均为金属原子top位。
由图4 中反应能垒ΔEbarrier可知,Cu 原子掺杂使Fe2O3载氧体反应能垒由2.30 eV 降低到1.68 eV(Cu原子top 位)和1.81 eV(Fe 原子top 位)。反应 能 垒降低,反应更容易发生。而且,由于Cu 原子存在,Cu-Fe2O3载氧体结构中Fe 原子top 位ΔEbarrier也降低。以上结果表明Cu-Fe2O3载氧体中Cu、Fe双金属元素之间具有互补和协同效应[33],使Cu-Fe2O3载氧体反应性显著提升。
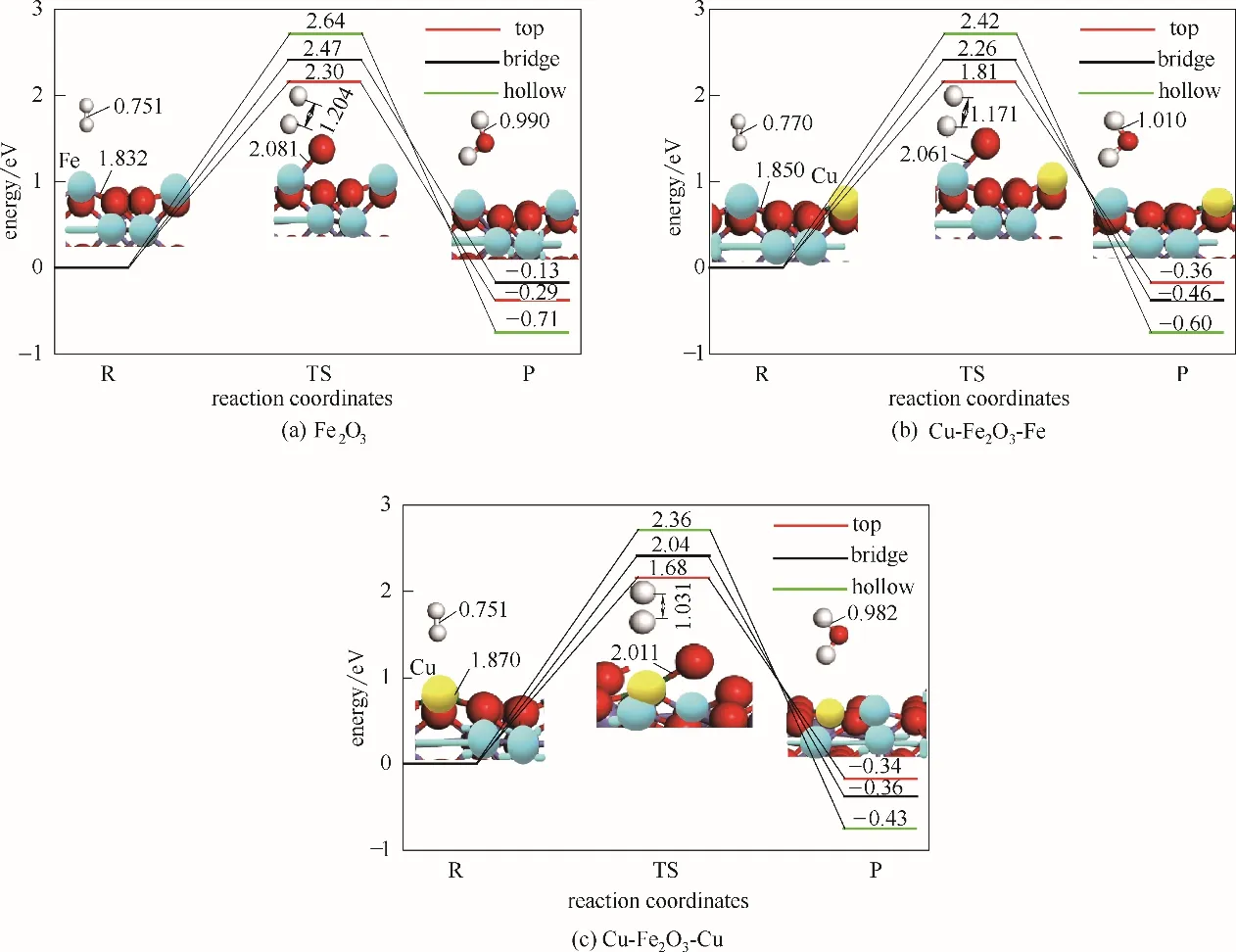
图4 载氧体与H2反应能垒图(键长单位Å):Fe2O3载氧体表面Fe原子位反应(a);Cu-Fe2O3载氧体表面Fe原子位反应(b);Cu-Fe2O3载氧体表面Cu原子位反应(c)Fig.4 Energy barrier diagram of oxygen carriers reaction with H2(band length unit:Å):reaction of Fe atom sites on Fe2O3oxygen carrier(a);reaction of Fe atom sites on Cu-Fe2O3oxygen carrier(b);reaction of Cu atom sites on Cu-Fe2O3oxygen carrier(c)
如图5 所示,H2在Cu-Fe2O3载氧体表面反应分两步进行:第一步,优先在Cu 原子位发生H2的氧化反应,生成H2O 从载氧体表面脱附,需要能垒1.68 eV。第二步,H2在Fe 原子位发生氧化反应,生成H2O 并从载氧体表面脱附,需要能垒1.81 eV。而单组分Fe-基载氧体表面只有Fe 原子位反应,即只发生Fe 原子位H2的氧化反应,需要能垒2.30 eV。通过微观反应机理研究表明:Cu-Fe2O3载氧体反应活性提升主要是因为Cu掺杂降低H2在Fe2O3载氧体表面能垒,进而改变Fe2O3载氧体反应路径。Cu 原子掺杂进入Fe2O3晶格后,使Fe2O3晶胞发生收缩(从5.036 Å 收缩到5.035 Å),并且Fe2O3载氧体表面部分Fe-O键(1.741 Å)和Fe-Fe键(3.150 Å)分别被Cu-O键(1.882 Å)和Cu-Fe 键(3.190 Å)取代,键长增大。表明键型、键长和晶胞大小均发生改变。相比Fe2O3载氧体,Cu-Fe2O3载氧体微观结构中,除了Cu 原子存在,Fe 原子周围化学环境(键型、键长和晶胞大小)也发生改变,使其与H2氧化反应能垒降低,反应活性增强。原单组分载氧体中只有Fe 原子位发生反应,Cu掺杂后转变为Cu和Fe原子位先后发生反应,且其反应能垒均低于单组分Fe-基载氧体。因此,低浓度Cu 掺杂Fe-基载氧体反应活性明显增强。该结果与实验获得的载氧体反应活性分析结果吻合,从微观角度阐释Cu-Fe2O3载氧体TGA 实验中反应活性提升的微观本质。
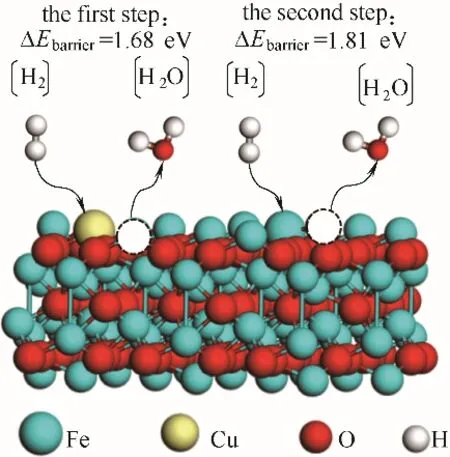
图5 H2在Cu-Fe2O3载氧体结构表面反应机理图Fig.5 Schematic diagram of Cu-Fe2O3oxygen carrier reaction with H2
图4 给出了三条最优反应路径。对反应物(R)-过渡态(TS)-产物(P)过程而言,首先是H2分子中H—H 键不断拉伸断键[以图4(a)Fe2O3为例,H—H:0.751-1.204-1.590 Å]。其次是载氧体表面与吸附位点最近的O 原子逐渐上移,使金属-氧键(Cu/Fe-O)不断拉伸断键[以图4(a)Fe2O3为例,Fe-O:1.832-2.081-2.580 Å],O 原子与已断键的2 个H 原子反应生成H2O[以图4(a)Fe2O3为例,H—O:3.410-1.941-0.990 Å],并从载氧体表面脱附。该过程中,H2在Fe2O3和Cu-Fe2O3载氧体表面的反应路径相似,主要是H2分子中H—H 键和载氧体表面金属-氧键(Fe/Cu-O)的断裂。然后反应生成H2O,从载氧体表面脱附。主要区别在于反应物、过渡态和产物结构中键型和键长不同,如图4所示,这是由载氧体微观结构不同所致。
需要指出的是,本文采用不同方法对Fe-基载氧体与H2反应过程进行研究,实验中测得表观活化能是从宏观角度对载氧体反应性能进行分析,其结果包括载氧体组成、微观结构和形貌等因素对其反应性能影响的综合结果。而DFT 计算是从分子反应过渡态的微观角度对载氧体与H2的基元反应机理进行研究。计算获得的ΔEbarrier与载氧体的微观结构有关,这些结构参数如原子种类、键型、键长等变化因素对其反应性能具有直接影响。虽然表观活化能和ΔEbarrier两者数据的确存在差别,但结果具有关联性。从实验和DFT 计算结果对比分析可知,Cu-Fe2O3载氧体性能提升主要是Cu 元素引入降低Fe2O3载氧体表面微观反应能垒,并使其反应路径改变,这是Cu-Fe2O3载氧体性能提升的根本原因。
3 结 论
本文通过TGA 实验和DFT 计算,对Cu-Fe2O3载氧体反应性能和微观机理进行研究,得到如下结论。
(1)TGA 实验结果表明,低浓度Cu 掺杂可将Fe2O3载氧体与H2反应的表观活化能由83.9 kJ/mol降低至72.3 kJ/mol。因此,低浓度Cu 的引入的确可提高Fe2O3载氧体晶格氧释放速率,进而增强了Fe-基载氧体的反应活性。
(2)通过DFT 理论计算阐明了低浓度Cu 掺杂Fe2O3载氧体与H2反应的微观机理。发现Cu掺杂使得Fe2O3载氧体表面反应能垒ΔEbarrier降低(从2.30 eV 降低至1.81 eV 和1.68 eV),改变了Fe2O3载氧体表面反应路径,使其反应活性明显增强。这与TGA实验分析结果吻合。
(3)由实验和DFT 计算结果的关联性可知,Cu-Fe2O3载氧体性能增强主要是Cu 元素引入降低微观反应能垒,并使Fe2O3载氧体微观反应路径改变。从实验角度为载氧体的筛选、优化和设计提供理论指导。
符 号 说 明
Ct——转化率,%
D——载氧体表面吸附H2分子到载氧体表面距离,Å
Ea——活化能,kJ/mol
EH2——H2分子优化后总能量,eV
Eoc——载氧体能量,eV
Eoc+H2——载氧体表面吸附H2分子后模型总能量,eV
EP——H2与载氧体表面O 反应生成产物H2O 后体系总能量,eV
ER——H2分子在载氧体表面稳定吸附后总能量,eV
ETS——反应过程中过渡态结构能量,eV
ΔEad——吸附能,eV
ΔEbarrier——反应能垒,eV
ΔEreaction——反应能,eV
P——产物
R——反应物
TS——过渡态
wi——起始样品质量,mg
wt——t 时刻样品质量,mg
wx——载氧体晶格氧完全释放后的样品质量,mg
