Nd掺杂CeO2(111)表面稳定结构的热力学分析实验设计
2020-11-17朱后禹刘东源郭文跃
朱后禹,刘东源,栾 波,赵 文,郭文跃
(1.中国石油大学(华东)材料科学与工程学院,山东青岛 266580;2.山东京博控股集团有限公司,山东滨州 262500)
随着科学技术和计算机的发展,科学研究的体系越来越复杂,传统的解析推导方法已不复应用,而计算机运算能力不断提高,为复杂体系的研究提供了新的科学手段—计算材料科学,并迅速得到发展。通过计算机模拟,可以初步了解不同温度、压强条件下材料的结构稳定性和使用性能等,基于所计算的材料性能演变规律和机理,可以为进一步改善材料性能提供理论指导。目前基于密度泛函理论(Density functional theory,DFT)[1]的Vienna ab initio simulation package (VASP),Material Studio 等软件所做的结构优化都是在0 K 温度下求解Kohn-Sham 方程[2](即电子密度泛函理论中的单电子方程)来求得体系的基态电荷密度,从而达到收敛标准。在很多应用上,比如准确地计算开壳层体系、具有高自旋多重度的体系、激发态体系等,还存在一定的局限性。因此,通过结构优化而得到的固体结构模型通常是满足化学计量数的无缺陷模型,而不是应用中某个温度压强下所处的一个实际状态。而用第一性原理计算热力学性质主要目的是预测固体材料的结构模型在某一温度和压强下处于一个什么样的实际状态。根据DFT计算得到结构模型的能量和熵,求出两个表面结构反应变化前后的吉布斯自由能变,再结合实际应用条件给定温度和压强数值,来判断这个结构处于什么样的相态。实验采用了基于密度泛函理论的第一性原理计算方法,依托于超算中心集群资源,通过模拟计算预测了掺杂CeO2材料在一定的温度和压强范围内表面氧空位的自发形成情况,从而确定实际反应条件下掺杂CeO2表面的稳定结构。
1 实验设计依据
CeO2是一种重要的稀土氧化物材料,在固体氧化物燃料电池、尾气处理和生物医药等方面都有重要的实际工业应用。大多数关于CeO2材料的研究都是利用其优越的氧化还原能力来做表面催化反应,具体表现在CeO2表面比较容易发生晶格氧原子的释放和迁移。在某一温度下,由于热涨落效应,氧原子会脱离晶格结构形成空位点缺陷,即氧空位。脱离的氧原子通常可以两两结合成为O2分子,或者直接参与表面催化氧化反应氧化吸附在表面的反应物。另外,晶格氧原子脱离形成一个氧空位后会在表面释放2个电子,由于Ce-4f 轨道的强局域性,电子会将近邻的Ce4+离子还原为Ce3+。由此可知,氧空位形成的数量具有饱和性,随着氧空位形成数量的增多,后续氧空位的形成越困难,CeO2对表面吸附物种的氧化能力会降低。因此,在做有关CeO2表面催化的模拟研究之前,有必要结合热力学计算预测实际反应条件下CeO2表面的氧空位形成情况。
近几年的研究都致力于对纯CeO2表面进行金属团簇修饰或金属原子掺杂来降低氧空位形成能,加速氧空位形成速率,从而提高CeO2表面的催化氧化反应速率。而实际应用较广的掺杂CeO2的氧空位形成能可调控至1~2eV。因此在中低温(300~800K)条件下,CeO2表面自发形成氧空位的可能性较大。在进行CeO2表面催化的模拟研究时,直接使用无缺陷的完整CeO2表面是不严谨的。本实验应用VASP 软件,以稀土金属元素Nd 掺杂CeO2表面为例,基于周期性密度泛函理论对Nd 掺杂CeO2进行结构优化,并通过热力学计算预测在高温还原性气氛条件下的表面氧空位形成情况,最终绘制Nd 掺杂CeO2表面稳定结构相图。
2 模型构建及参数设置
采用Vienna Ab-into Simulation Package(VASP)计算软件,并基于超算中心集群开展计算工作。
2.1 模型构建可分为如下几个步骤
(1)从Material Studio 软件自带的结构库中导入CeO2的晶胞结构;
(2)利用建模工具进行切面操作得到CeO2(111)面,建立超晶胞,设置真空层高度;
(3)用Nd 原子替换CeO2(111)表面的一个Ce 原子,期间需充分考虑Nd 原子在表面上各种可能的替位掺杂位置,并通过结构优化比较不同掺杂结构的总能量,确定最稳定的Nd 掺杂CeO2(111)表面结构。
如图1所示,Nd 掺杂CeO2(111)表面模型为六层原子组成,每层含有9个原子(3×3)的超晶胞模型。在表面垂直方向添加了15nm 的真空层,以防止在该方向上出现周期性结构的相互作用。在模拟过程中,对Nd 掺杂CeO2(111)表面模型的上三层原子进行无对称约束的结构优化,并固定下三层原子于计算的体相点阵位置,以模拟固体内部几乎不受表面弛豫影响的情况。
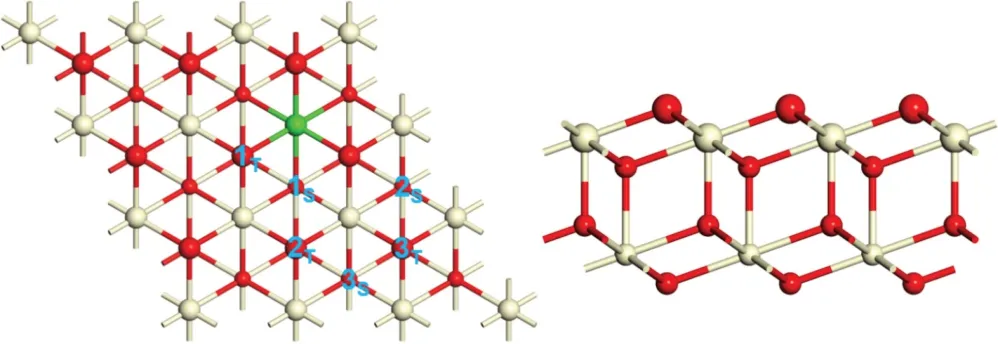
图1 Nd掺杂CeO2(111)表面模型的俯视图(左)和侧视图(右)。为了加以区分,最顶层的两层原子用大号球棍模型显示。红色和浅黄色表示O原子和Ce原子,其中一个Ce原子被Nd原子取代并以绿色标示。图标O1T-O3T和O1S-O3S分别表示表层和次表层可能形成的氧空位位置。
2.2 计算参数设置如下
所有计算都是在以DFT 理论为基础的VASP 软件中进行的。离子-电子间相互作用采用平面波增强方法(PAW)处理,电子间交换关联势能采用广义梯度近似(GGA)处理。在静态自洽计算中,Ce(5s,5p,6s,4f,5d),Nd(5s,5p,6s,4f,5d),和O(2s,2p)作为价电子处理,其余电子与原子核视为离子并用PBE 赝势代替。在所有结构优化中设置截断能为400eV,离子位移收敛标准为0.03eV/Å,电子能量收敛标准为10-6eV。所有计算均采用自旋极化。布里渊区内的积分采用3×3×1的Monkhorst-Pack k 点设置。采用DFT+U 方法处理Ce-4f 轨道的强关联相互作用,其中Hubbard U修正数值U-J设置为5eV。
氧空位形成能(Evac)计算公式是:
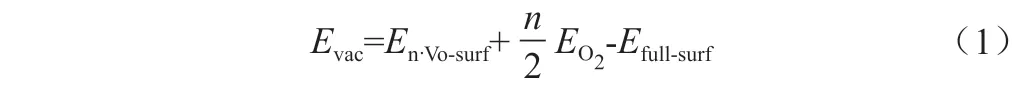
其中,En·Vo-surf,EO2和Efull-surf分别表示生成n个氧空位后的Nd 掺杂CeO2(111)表面的总能量,真空中一个氧气分子的能量以及无缺陷表面的总能量。从头算热力学的基本原理是把Nd 掺杂CeO2(111)表面生成一个氧空位的过程等价于一个固体表面被还原性气体还原的具体反应过程,从而与温度和还原性气体的分压建立函数关系,计算该氧化还原反应的吉布斯自由能变。具体反应表达式可用如下式子表示
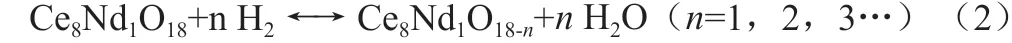
其中n表示在一个反应周期内有多少H2分子被Nd 掺杂CeO2(111)表面氧化,同时生成了n个氧空位以及最终产物H2O。则该反应过程的吉布斯自由能变可用如下式子表示:
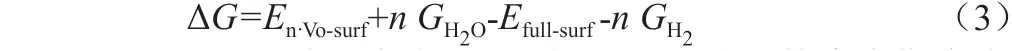
GH2OandGH2表示气相H2O 分子和H2分子的吉布斯自由能。单位摩尔的气相分子H2O 或H2的吉布斯自由能可进一步由两者的化学势μH2O(T,P)和μH2(T,P)表示:
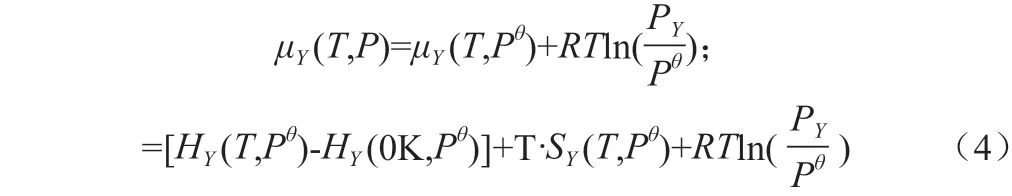
式中,下标Y表示气相分子H2O 或H2,R为热力学理想气体常数8.314J/(mol·K),标准热力学数据焓HY(T,Pθ)和熵SY(T,Pθ)可从NIST 官方数据库中查询。结合式(3)、(4)可知,当ΔG=0,n=1时,可得到压强pY与温度T的函数关系曲线,此曲线的物理意义即为无缺陷的Nd 掺杂CeO2(111)表面和生成一个最近邻氧空位表面的临界状态。以此类推,代入n=2,3…时可得到形成后续氧空位的临界相态曲线。以压强pY与温度T为坐标,整合n=1,2,3…代入后得到的临界相态曲线即可。
3 实验结果及讨论
纯CeO2(111)表面的表层氧空位形成能约为2.7eV,比贵金属氧化物更易于形成氧空位,但仍不能满足固体氧化物燃料电池启动温度低温化的需求,所以本研究中也采用当前研究较为广泛的稀土金属Nd 掺杂CeO2模型,来进一步调控氧空位形成能。图2为结构优化并最终达到收敛标准的Nd 掺杂CeO2(111)表面模型,其中一个表层金属Ce 原子替换为Nd原子。首先,由于掺杂原子引入,导致了一定程度上的晶格畸变。相比于纯CeO2(111)表面中的Ce-OT,Ce-OS 键长2.355Å和2.356Å,Nd 原子与表层晶格氧原子OT之间的键长拉伸至为2.362Å,与次表层氧原子OS之间的键长缩短为2.348Å。Nd掺杂破坏了晶格结构原有的对称性,利于表面氧空位的形成。
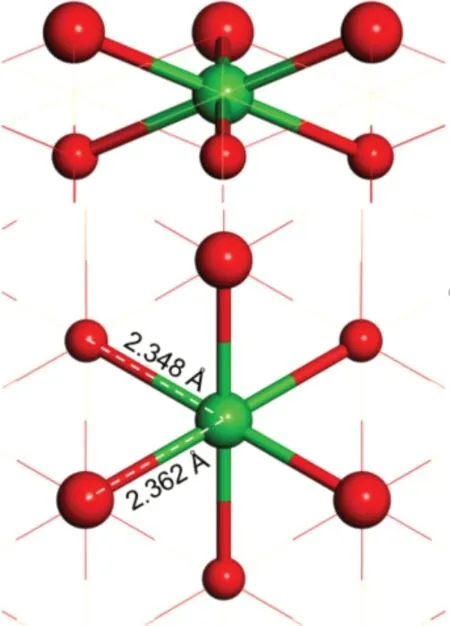
图2 Nd掺杂CeO2(111)表面的优化结构及相关键长参数
根据周围晶格氧原子与掺杂原子距离的不同,需要考虑6个可能的氧空位形成位置。计算其氧空位形成能(表1)后发现,相对于掺杂原子,最近邻氧原子脱离晶格形成氧空位所需能量最低,只有2.253eV。而最近邻次表层氧空位形成能为2.483eV。这一结果与上文讨论的键长数据相吻合,氧空位形成能的大小与金属-氧原子键长呈正相关。另外,从整体规律上来看,距离掺杂原子越远,氧空位形成能越大,趋向于纯CeO2(111)表面的2.7eV。由表1所示数据推断,在催化氧化表面分子的反应过程中,与掺杂原子最近邻的表层氧原子会优先被释放,形成氧空位。

表1 Nd掺杂CeO2(111)表面不同位置的氧空位形成能
如上文所述,从NIST 查得的标准热力学数据焓HY(T,Pθ)和熵SY(T,Pθ)是在某些特定温度和标准压强下测定的离散数值,不能满足表面结构相图所需的连续曲线关系。但根据理想气体的热力学关系,可用Origin 进行数据拟合使之成为关于温度的连续函数H(T)=E+nRT和S(T)=CplnT-Rlnp+S0。拟合结果如图3所示。
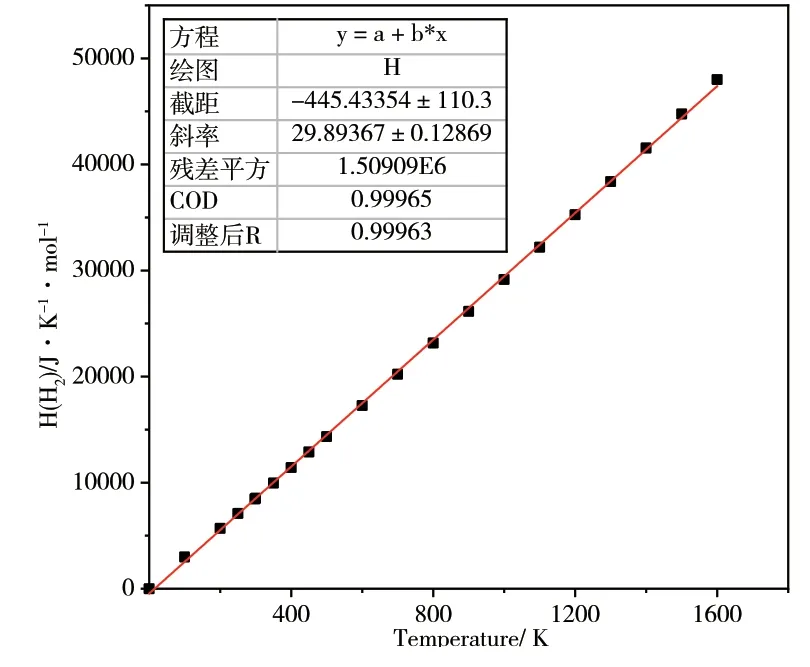
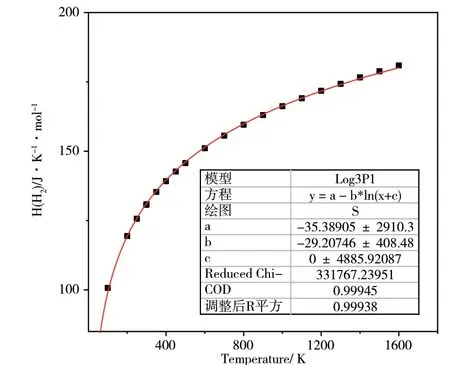
图3 实验反应温度范围内气相H2分子的标准热力学数据焓HH2(T,Pθ)和熵SH2(T,Pθ)的拟合曲线
因扩散速率通常比化学催化反应速率快几个数量级,所以当固体氧化物燃料电池阳极催化反应经过一段时间达到平衡时,最终产物H2O 的生成速率也会迅速趋于稳定,另外,燃料电池运行过程中产生的燃料废气会及时向外界排出,所以气相中H2O 的分压也会迅速趋于一个定值,具体可参考实验中的气相组分测量值。而通入的反应气体H2可以人为调控,所以H2的气体分压是气相环境的决定因素。按照3.2节中所述方法代入数据处理,可得到以H2分压PH2和温度T 为横纵坐标的热力学稳定结构相图,如图4所示。在固体氧化物燃料电池阳极的实际反应条件下(黑色虚线标示,PH2=1atm,T=800K),一个氧原子会脱离晶格形成氧空位,即Nd 掺杂CeO2(111)表面可以提供一个晶格O 原子催化氧化H2分子。由此不难推断,在上述反应条件下,体系的初始结构为H2吸附在无缺陷的Nd 掺杂CeO2(111)表面。经过一系列基元反应之后,最终H2被Nd 掺杂CeO2(111)表面氧化为H2O 分子并在表层留下一个氧空位。即确定了Nd 掺杂CeO2(111)表面催化氧化H2反应这一体系的初末态,为后期研究Nd 掺杂CeO2(111)表面的H2催化氧化反应机理奠定了较为准确的模型基础。此外,也说明了Nd 掺杂CeO2(111)表面在此条件下是稳定的晶格结构,晶格中的Nd 掺杂原子仍然保持较为稳定的6个O 原子配位环境。只有当温度过高,反应气体中H2分压较大时,才可能使表面形成3个以上的表层氧空位,金属离子配位数急剧降低,从而发生原子聚集和Ostwald 效应,破坏表面的稳定性。
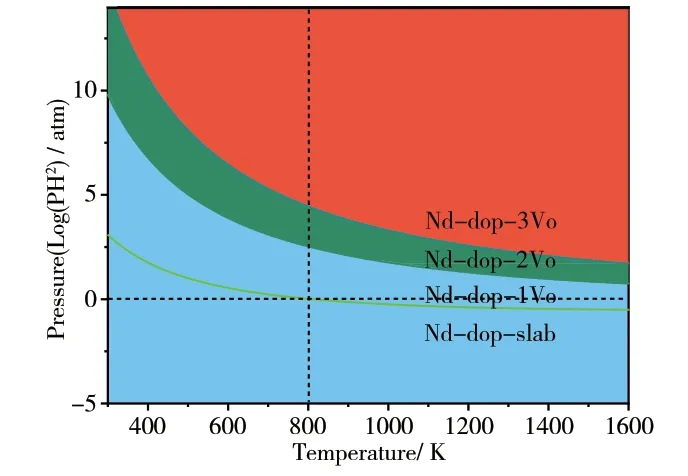
图4 反应条件下Nd掺杂CeO2(111)表面相图
不同的颜色区域表示在该温度和压强环境下的稳定结构。固体氧化物燃料电池阳极的反应条件用虚线标示(PH2=1atm,T=800K)。
4 结论
本实验设计依托超算中心计算平台,采用VASP 软件,并结合从头算热力学方法研究了反应条件下Nd 掺杂CeO2(111)的表面相态,从微观角度研究了掺杂CeO2表面结构随温度和压强的变化,确定了反应条件下的Nd 掺杂CeO2表面稳定结构。结合从头算热力学绘制表面结构相图的方法可以提供实验条件下表面结构的热稳定性,并推测不同温度和压力条件下的表面状态及构型。
