MoS2-壳聚糖气凝胶原位还原金硫代硫酸根络合离子的基础研究
2020-11-14孙凯歌贾菲菲
孙凯歌 陈 鹏 贾菲菲
(武汉理工大学资源与环境工程学院,湖北武汉430070)
金作为贵金属的代表具有重要的政治、经济、社会价值。氰化法提金是最为古老、使用最为成熟的技术,其具有试剂消耗量少、金的回收率高等优点,当今世界约92%的金矿均采用氰化法提取[1]。然而,氰化物具有剧毒性,对环境和生态造成严重污染和破坏;此外,氰化物对一些含铜、砷、碳等元素的复杂金矿的处理效果较差,技术上也限制了氰化法的继续使用[2,3]。为此,寻找高效、无毒的氰化物替代品迫在眉睫。硫代硫酸盐法提金不仅具有浸出速度快、无毒无害、成本低廉等优点,而且能够有效处理含铜含碳等氰化法无法有效浸出的复杂金矿,因此,硫代硫酸盐被认为是最有可能替代氰化物的无毒无害浸金试剂[4,5]。然而,硫代硫酸盐提金工艺至今没有实现工业化应用,其主要难点之一为浸出后的金络合离子(Au(S2O3)23-)回收困难。目前,Au(S2O3)23-的回收方法被广泛研究,其中主要包括置换法、溶剂萃取法、电沉积法和吸附法[6-9]。置换法、溶剂萃取法和电沉积法存在较大的限制性,其对金浓度要求较高、成本高,不适用于浸出液中金的提取;吸附法主要包括离子交换树脂和活性炭吸附。离子交换树脂在吸附过程中矿浆需要过滤,成本较高,树脂本身易粉化且不易再生;活性炭具有良好的吸附性能,被广泛应用于氰化提金工艺中,但活性炭、改性活性炭等对Au(S2O3)23-吸附能力极弱,因此硫代硫酸盐法不能像氰化法一样形成“炭浆法”工艺来处理金矿石。此外,目前黄金回收工艺较为复杂,包括吸附、脱附和还原等步骤。因此,开发一种不仅能够有效回收Au(S2O3)23-而且直接将Au(S2O3)23-还原获得金单质的方法具有重要意义。
二硫化钼(MoS2)是由两层硫原子夹1层钼原子构成的层状过渡金属硫属化合物[10]。MoS2纳米片具有巨大的理论比表面积、优良的吸附性能以及优异的光催化还原性能等,已被广泛应用于多个领域。研究发现Au(S2O3)23-/Au0的还原电位为 0.15 eV[11],而MoS2的导带位置约为0 eV,较Au(S2O3)23-/Au0的还原电位更负,因此,MoS2易实现将 Au(S2O3)23-还原为Au0。同时,由于S原子对Au等贵金属具有强烈的亲和作用力,表面富含S原子的MoS2对Au(S2O3)23-具有良好的络合能力。ZHAN等[12]深入研究了MoS2/ZnS纳米异质结对 Au(S2O3)23-的原位光催化还原性能,结果表明MoS2/ZnS纳米异质结能够将Au(S2O3)23-还原为金纳米颗粒,其还原量高达1 120.56 mg/g,其还原机理为MoS2/ZnS被光激发后产生大量的光生电子将 Au(S2O3)23-还原。因此,MoS2基光催化剂对 Au(S2O3)23-具有优异的回收性能,然而,MoS2纳米材料极难实现固液分离,在实际生产中难以应用。
因此,本文旨在通过MoS2纳米片三维化,制备易固液分离的三维多孔MoS2-壳聚糖气凝胶(MoS2-CSA),深入探究MoS2-CSA催化还原Au(S2O3)23-的性能和机理。
1 试验原料与试验方法
1.1 试验原料
壳聚糖(CS)、钼酸钠(Na2MoO4·2H2O)、硫代乙酰胺(CH3CSNH2)、硫代硫酸铵(H8N2O3S2)、戊二醛(C5H8O2)、乙酸(C2H4O2)、氢氧化钠(NaOH)以及乙醇(C2H6O)均为分析纯试剂,购买自国药集团化学试剂公司。金标液(HAuCl4)购买自美国加联仪器有限公司。
1.2 试验仪器
试验采用德国布鲁克公司的X射线衍射仪获得样品的物相及结构特征,Vector-22型红外光谱获得样品的官能团信息,采用日本电子株式会社的JSMIT300型场发射扫描电子显微镜对样品的形貌进行观测,采用日本岛津公司的AA-6880型原子吸收光谱检测溶液中金的浓度。
2 试验方法
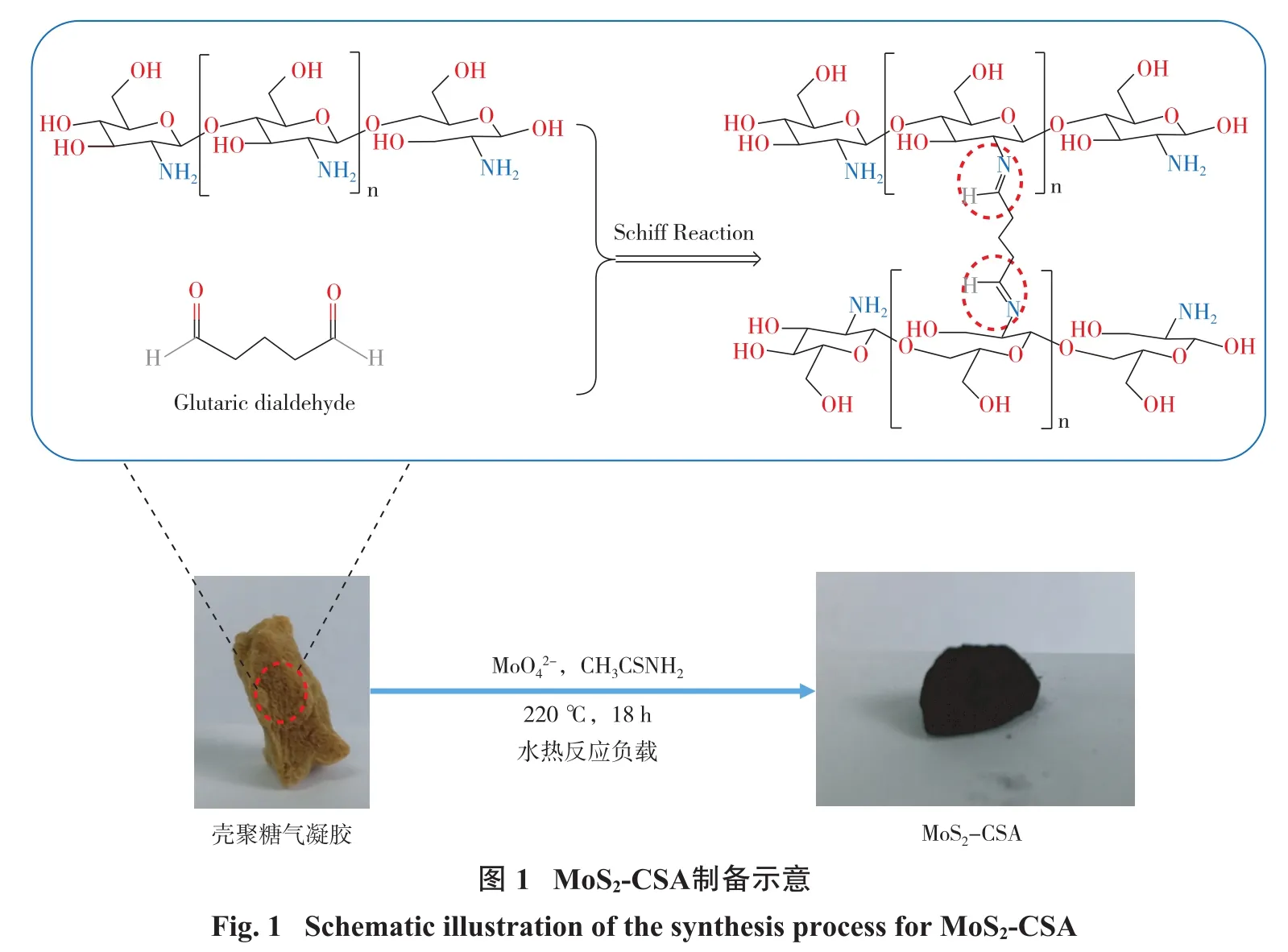
2.1 MoS2-壳聚糖气凝胶(MoS2-CSA)的制备
首先,称取0.3 g壳聚糖(CS)加入到醋酸质量分数为2.5%的20 mL溶液中,在转速为500 r/min下持续搅拌直至形成均匀溶液;再取1.5 mL戊二醛,加入到上述均一溶液中,继续搅拌2 min,CS与戊二醛发生Schiff交联作用,形成CS凝胶。将此凝胶于冰箱冷冻1 h后,再放入真空冷冻干燥机冷冻12 h,即得壳聚糖气凝胶(CSA)[13]。继而,称取 180 mg Na2MoO4·2H2O和360 mg CH3CSNH2加入到80 mL去离子水中,以转速500 r/min持续搅拌约30 min至其完全溶解后,将80 mg上述制备的CSA加入其中,继续搅拌30 min。将混合物倒入100 mL的聚四氟乙烯内胆中,将该内胆放入增压釜中,在220℃马弗炉中反应18 h。将反应物分别用去离子水和乙醇多次洗涤后,放入真空冷冻干燥机,冻干至恒重,即得MoS2-CSA。制备示意图如图1所示。
2.2 Au(S2O3)23-的配置
取200 mL去离子水,调节其pH为10.0,然后向其中加入0.51 mmoL的硫代硫酸铵,在400 r/min转速下搅拌使其溶解得到硫代硫酸根(S2O32-)溶液。控制pH不低于10,向200 mL硫代硫酸根溶液中逐滴加入25 mL金标液(1 g/L,即0.126 mmol),金标液滴加完后,保证溶液pH为10,然后转入250 mL容量瓶中加水定容至刻度线,得到含金量为100 mg/L的 Au(S2O3)23-溶液。其他浓度的 Au(S2O3)23-的配制方法与此相同。
2.3 Au(S2O3)23-的还原
将10 mg MoS2-CSA光催化剂加入到初始pH值为10、体积为150 mL、金含量为100 mg/L的Au(S2O3)23-溶液中。在25℃室温环境下,以180 r/min的转速振荡,一定时间间隔后取1~2 mL溶液,经孔径为0.22 μm滤膜过滤并稀释后用原子吸收光谱测试滤液中金的残余浓度。溶液初始pH影响试验中,用NaOH和 HCl将 Au(S2O3)23-溶液的初始pH值调节为8~11,将10 mg MoS2-CSA光催化剂加入到浓度为100 mg/L的 Au(S2O3)23-溶液中。对于金初始浓度影响试验,Au(S2O3)23-的初始浓度范围为 15~200 mg/L,10 mg MoS2-CSA光催化剂加入到初始pH值为10,体积为150 mL的Au(S2O3)23-溶液中。
Au(S2O3)23-的还原量通过以下公式计算得出:
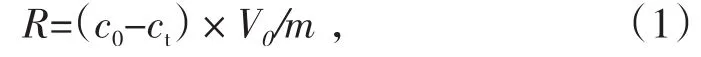
式中,R为Au(S2O3)23-的还原量,mg/g;c0和ct分别为初始溶液和t时刻Au(S2O3)23-的浓度,mg/L;m为催化剂的质量,g;V0为溶液的体积,L。
3 试验结果及分析
3.1 MoS2-CSA光催化剂的表征
图2为纯MoS2纳米片以及MoS2-壳聚糖气凝胶(MoS2-CSA)的XRD图谱。纯MoS2纳米片以及MoS2-CSA的衍射峰的位置一致,表明MoS2负载到壳聚糖凝胶上时,结构未发生改变。衍射峰在2θ为32.8°、35.5°以及 57.0°分别对应 MoS2的(100)、(103)和(110)面[14],与之前的文献报道一致,说明水热反应成功制备了MoS2纳米片。本应位于衍射角为14.3°的(002)面的衍射峰消失不见,取而代之的是衍射角分别位于9.4°和17.0°的特征峰,分别被标记为(001)*和(002)*,这可能是由于MoS2-CSA中MoS2层间距较纯MoS2纳米片增加所致[15]。从致密态到膨胀态层状结构的转变有利于MoS2纳米片提高光催化活性。相较于纯MoS2纳米片,MoS2-CSA的峰强度降低,说明MoS2-CSA上的MoS2纳米片堆叠减少,片层数更少,活性更高。
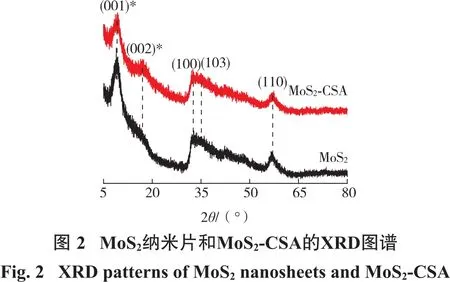
图3为MoS2、CSA以及MoS2-CSA的FTIR图谱。对于纯MoS2纳米片,位于606 cm-1的峰为Mo—S键的振动峰;位于1 396 cm-1处的吸收峰为S…H之间的氢键作用;3 452 cm-1处的吸收峰为MoS2表面吸附水的—OH振动峰[16];对于CSA,位于1 638 cm-1处的吸收峰为亚胺键(HC=N—);1 713 cm-1处的吸收峰为酰胺I(—NH2,伯酰胺)的拉伸振动峰;位于2 940 cm-1处的吸收峰是由于壳聚糖中C—H键的伸缩振动产生的;位于3 423 cm-1处的吸收峰为—NH和—OH的重叠的伸缩振动峰[17];在MoS2-CSA中,MoS2与CSA的特征吸收峰均被发现(611 cm-1,1 625 cm-1,1 713 cm-1,2 929 cm-1以及 3 442 cm-1),这表明 MoS2与 CSA成功复合到一起。
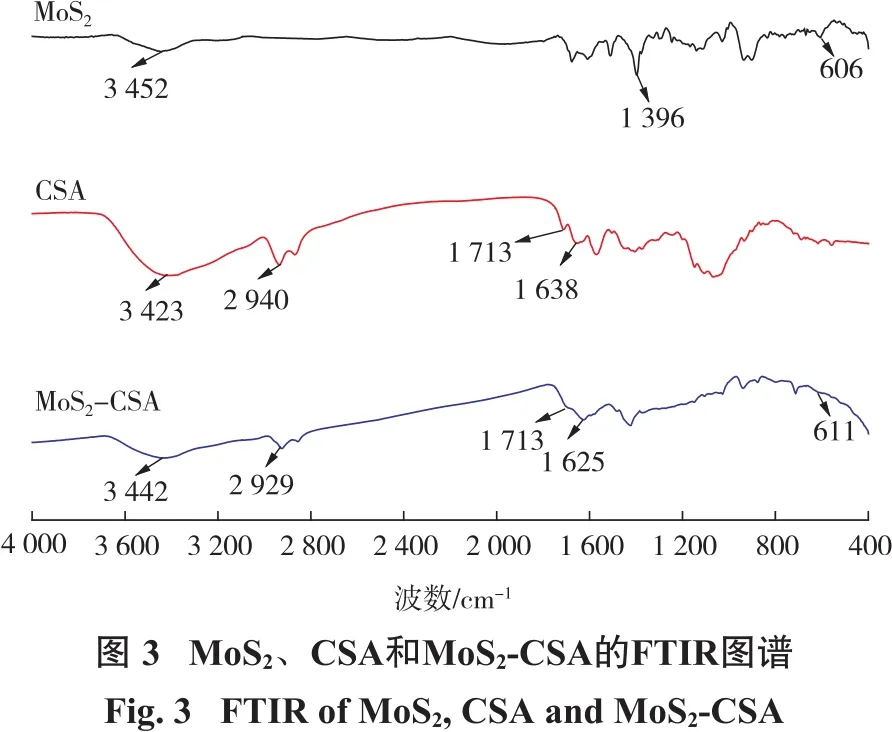
图4为壳聚糖气凝胶与MoS2-CSA的SEM图。从图4(a)可以看出,壳聚糖(CS)与戊二醛(GA)发生Schiff交联反应生成的CSA呈现出丰富的孔洞结构,孔道之间相互贯穿连通,有利于MoS2的附着和溶液中离子的扩散。从图4(b)中观测到MoS2-CSA亦具有丰富的孔洞结构。图4(c)(d)中观察到纳米片状的MoS2均匀地负载到CSA表面。MoS2-CSA提升了材料比表面积,暴露了更多催化活性位点。此外,块状凝胶结构利于反应后的固液分离,克服了纳米材料在液相中难以分离的难题。
3.2 Au(S2O3)23-的催化还原
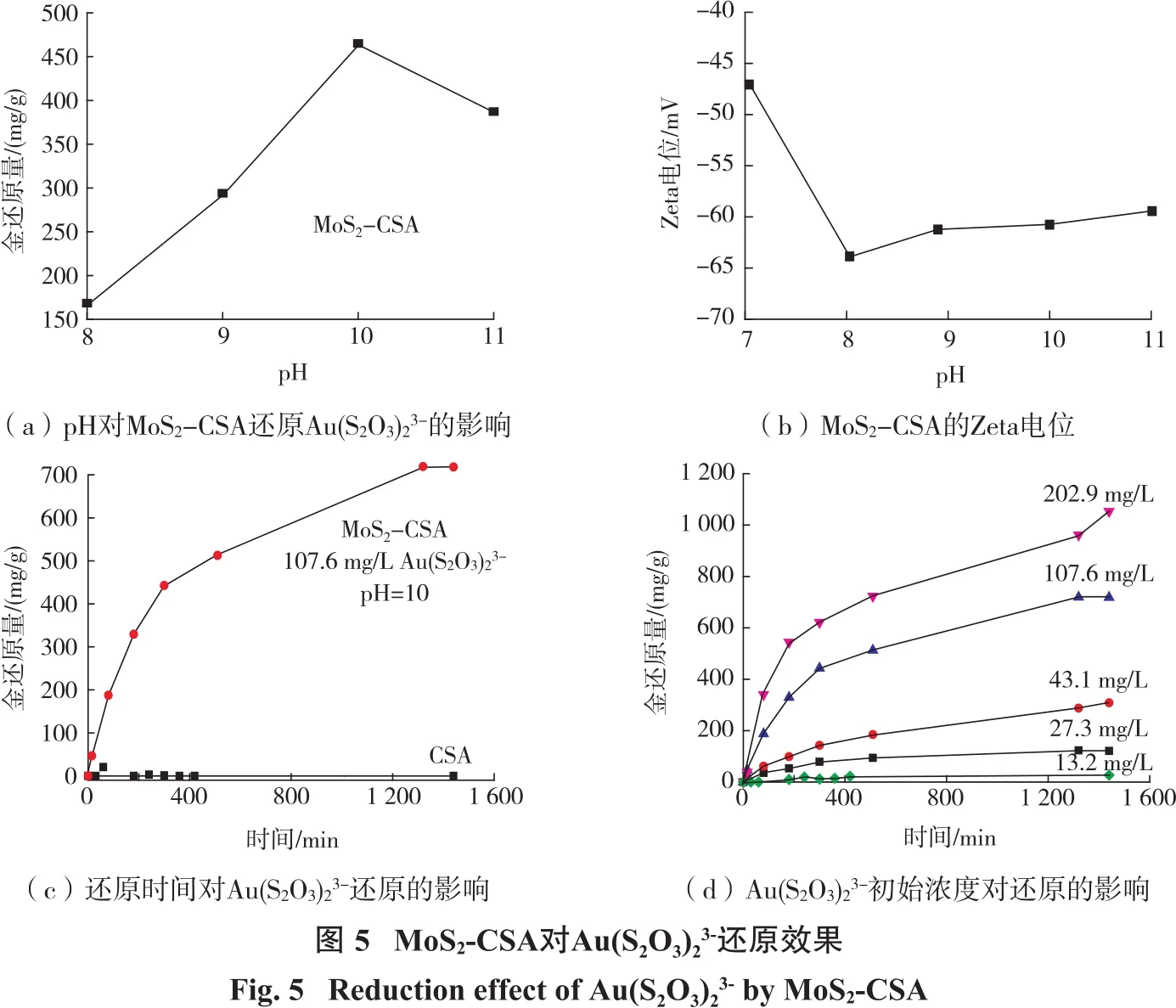
图5(a)为溶液pH对MoS-CSA还原Au(SO)3-2232(约52 mg/L)的影响,MoS2-CSA的Zeta电位随pH的变化趋势见图5(b),图5(c)为MoS2-CSA与CSA对Au(S2O3)23-初始浓度为107.6 mg/L、pH为10时的催化还原性能,图 5(d)为不同 Au(S2O3)23-的初始浓度下MoS2-CSA 对 Au(S2O3)23-的还原性能的影响。
如图 5(a)所示:随 pH 升高,MoS2-CSA对Au(S2O3)23-的还原量逐渐增加;当pH到达10时,MoS2-CSA 对 Au(S2O3)23-的还原量基本达到最大,当pH超过10后,Au(S2O3)23-的还原量逐渐下降。从图5(b)看出:在整个测试pH范围内,MoS2-CSA表面均表现出很强的负电性(均低于-40 mV),且pH升高时MoS2-CSA表面负电性呈现先增强后趋于稳定的趋势。阴离子Au(S2O3)23-与强负电性的MoS2-CSA呈现强烈的相互排斥作用,因此,Au(S2O3)23-通过静电作用吸附在MoS2-CSA表面是非常困难的。MoS2-CSA对 Au(S2O3)23-优异的回收性能归功于MoS2的光催化还原能力。从图5(c)可知:MoS2-CSA对Au(S2O3)23-具有优异的回收能力;随着反应时间的增加,金的还原量逐渐增加,最终达到平衡,其最大还原量高达718.3 mg/g;而CSA 对Au(S2O3)23-无回收能力。从图5(d)可以看出,随着溶液中Au(S2O3)23-初始浓度的增加,MoS2-CSA 对 Au(S2O3)23-的还原量逐渐增加。一般而言,溶液中 Au(S2O3)23-浓度增加,MoS2-CSA与 Au(S2O3)23-之间的碰撞接触频率增加,更多的Au(S2O3)23-离子靠近接触MoS2-CSA的表面,从而更多的 Au(S2O3)23-可以被MoS2-CSA还原。

3.3 还原机理分析
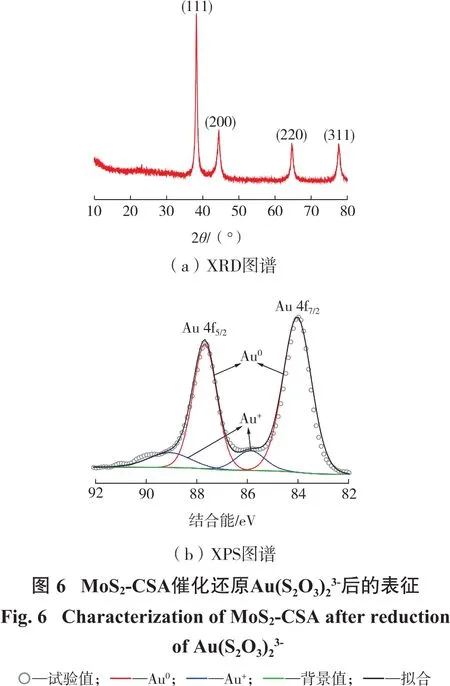
为了验证MoS2-CSA对金的大量回收是由于Au(S2O3)23-被还原为金单质,对反应后的MoS2-CSA样品进行了XRD分析和XPS分析,见图6。
从图6可以看出:在MoS2-CSA催化还原Au(S2O3)23-后,在衍射角 2θ为 38.2°、44.4°、64.7°以及77.7°出现了明显的特征峰,根据JCPDS04-0784标准卡片可知,这为金单质的特征衍射峰,其分别对应金单质的(111)、(200)、(220)和(311)面,这证明了 Au(S2O3)23-被 MoS2-CSA 还原成了金单质[18]。对Au 4f轨道的XPS图谱进行分峰拟合,位于83.98 eV和87.68 eV处两个较高的特征峰分别对应金单质的 Au 4f7/2和 Au 4f5/2,说明大量的Au(I)被还原为金单质;位于85.88 eV和89.08 eV处两个小峰属于Au(I)的特征峰,这可能由于MoS2-CSA大量的孔洞结构,有利于溶液中离子的扩散,导致其吸附了少量的 Au(S2O3)23-离子[19]。
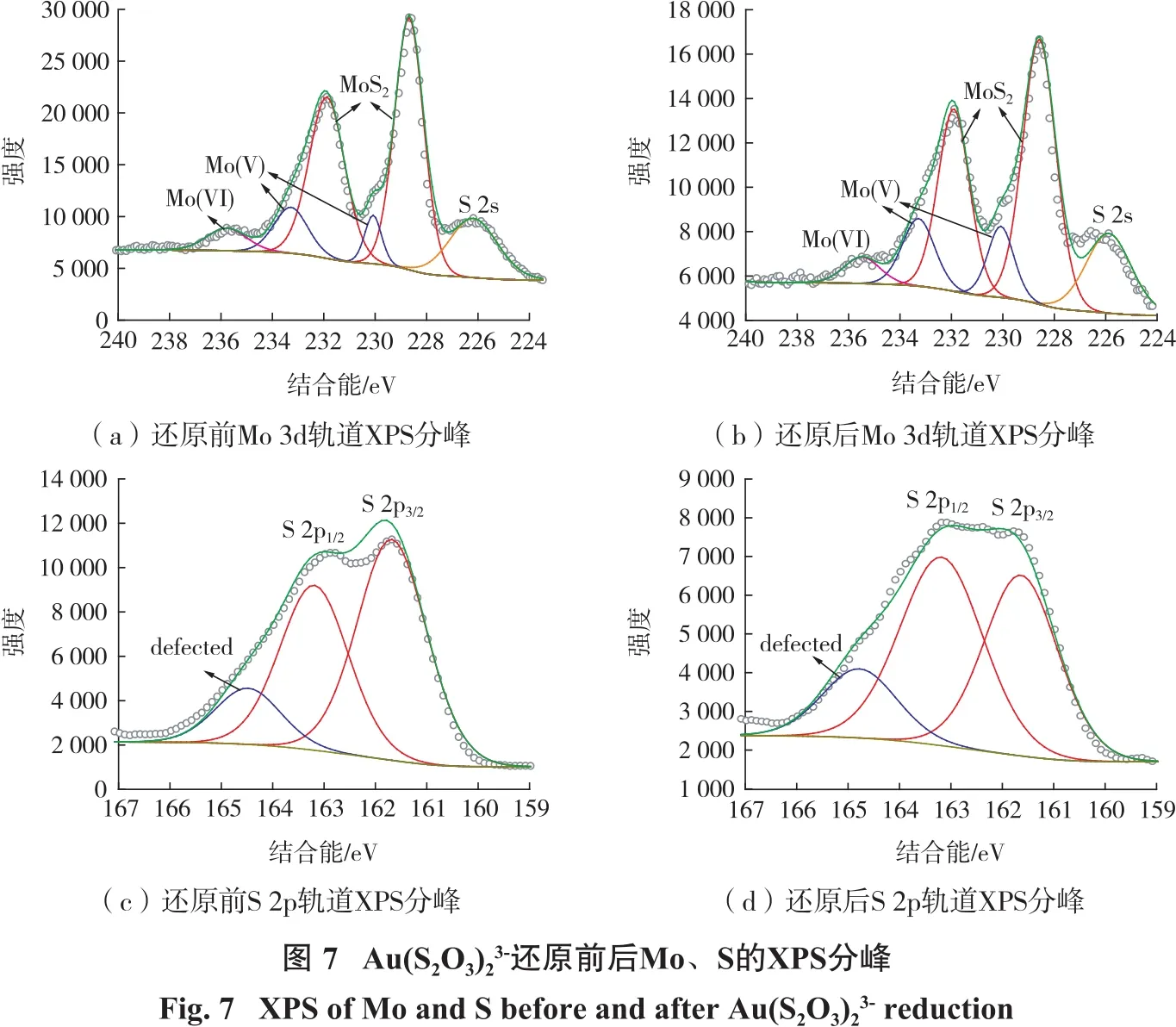
为了进一步探究 Au(S2O3)23-的还原机理,对与Au(S2O3)23-反应前后的MoS2-CSA的Mo、S的XPS窄谱进行了分峰拟合,见图7。
从图7(a)可以看出:反应前Mo 3d轨道的XPS图谱中,结合能位于228.68 eV和231.88 eV的峰为MoS2中Mo(IV)的化学相态;结合能位于226.18 eV的峰为MoS2中S2-的特征峰;位于230.08 eV和233.28 eV的峰为Mo(V)的特征峰;结合能位于235.78 eV的特征峰属于Mo(VI)的特征峰;由上可知,Mo主要以MoS2的形态存在,说明成功制备了 MoS2纳米片[20,21];少量的 Mo(V)和 Mo(VI)分别以 Mo2S5和 MoS3的化学形态存在,这可能是由于在水热合成二硫化钼过程中,钼源、硫源反应不够充分所致。图7(b)中,在还原 Au(S2O3)23-后,Mo(IV)、Mo(V)以及 Mo(VI)的物相组成没有发生明显变化,但是结合能整体出现了向右偏移,这是因为金纳米颗粒附着在MoS2表面导致的。对于反应前S 2p轨道而言(图7(c)),结合能位于161.68 eV和163.19 eV的峰属于MoS2的峰;位于164.48 eV的峰为缺陷相,是由于未完全反应的水热反应导致的;在还原Au(S2O3)23-后,代表 MoS2中 S的分峰未发生明显变化,分别位于161.64 eV和163.18 eV,而缺陷相的峰结合能变大,左移至164.78 eV,这可能是由于MoS2-CSA丰富的孔洞结构导致少许 Au(S2O3)23-被MoS2所吸附,MoS2中S缺陷与Au(S2O3)23-相互作用导致结合能变大,该结果也说明了图7(b)中Au(I)的来源。
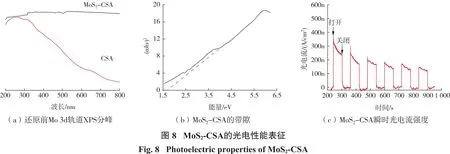
MoS2优异的光催化性质是其可以还原Au(S2O3)23-的根本原因,因此,对MoS2-CSA进行了光电性质分析。图8(a)为MoS2-CSA与CSA的光吸收性能分析,CSA只对能量较高的紫外光有吸收性能,而对可见光利用较少;MoS2-CSA在整个紫外-可见光(200~800 nm)范围内都具有良好的吸收性能,这得益于所负载的MoS2纳米片对光具有良好的吸收性能。半导体的带隙对催化作用有至关重要的影响,带隙能可由以下公式(2)对数据进行处理[22]:
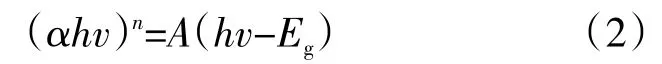
式中,α、h、v、Eg和A分别代表吸收系数、普朗克常数、光频率、带隙能和常数。n取决于半导体的性质,对于直接带隙半导体n取2,对于间接带隙半导体n取1/2,MoS2为直接带隙,因此n取2。以(αhv)n为纵坐标,hv为横坐标作图得图8(b),其切线与横坐标交点即为带隙能。根据图8(b)分析得其带隙能为1.7 eV,较窄的带隙能利于MoS2-CSA充分吸收利用太阳光。进一步,MoS2-CSA的导带和价带的位置可以由式(3)和(4)计算得出[23]:

式中,X为半导体的绝对电负性,Ee为氢的自由电子能(4.5 eV),EVB和ECB分别为半导体的价带和导带位置,Eg为半导体的带隙能(由图8(b)得)。因此,MoS2-CSA的ECB和EVB分别为-0.03 eV、1.67 eV。其导带位置较 Au(S2O3)23-/Au0的还原电势0.15 eV更负,能够将Au(S2O3)23-还原为金纳米颗粒。瞬时光电流是一种研究半导体电极/溶液界面光生电荷转移特性的有效方法。图8(c)为MoS2-CSA的瞬时光电流响应。由图8(c)可以看出,当MoS2-CSA受到光激发时,产生了较强的电流响应,表明MoS2-CSA能够有效产生并传输光生电荷,具有较强的光催化还原能力。
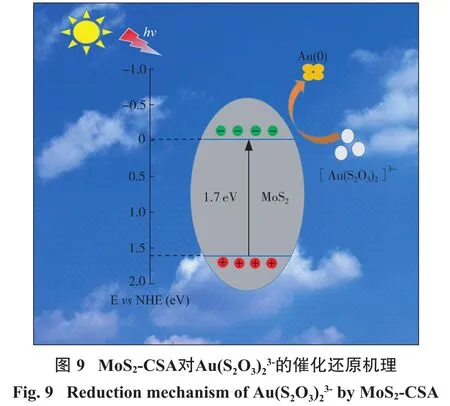
基于以上分析结果,MoS2-CSA光催化还原Au(S2O3)23-的机制可以总结为如下图9所示过程。由于 MoS2中 S 原子与 Au(S2O3)23-较强的亲和作用,Au(S2O3)23-被络合到MoS2表面,当MoS2-CSA被光激发后,MoS2的导带上产生光生电子,然后,光生电子将络合到表面的Au(S2O3)23-原位还原为金纳米颗粒,实现金的一步回收。
4 结 论
通过简易负载方式制备了三维多孔结构的MoS2-壳聚糖气凝胶(MoS2-CSA),MoS2-CSA具有较大的宏观结构,能够轻易实现固液分离。金络合离子回收试验表明,MoS2-CSA对Au(S2O3)23-不仅具有优异的回收性能,而且将Au(S2O3)23-原位还原为金单质。机理研究表明MoS2-CSA的光催化还原在Au(S2O3)23-的还原回收中起到关键作用。研究结果对硫代硫酸盐提金的发展具有重要意义。
