miR-199a在LPS诱导的大鼠原代心肌细胞中通过激活NF-κB信号通路加重心肌细胞损伤
2020-11-09可董平栓和素娜陈士芳赵希坤张恒亮王腾飞
王 可董平栓*和素娜陈士芳赵希坤张恒亮王腾飞
(1.河南科技大学第一附属医院,河南洛阳 471003; 2.河南科技大学医学院,河南 洛阳 471003)
心肌炎是一种炎症性心肌病,其特征主要表现在心肌细胞的水肿,变性和坏死[1]。心肌病变时出现广泛的炎症浸润和氧化应激反应,最终导致恶性心律失常、心源性休克甚至心脏骤停。从病原微生物释放的许多内源性和外源性因子,如自身抗原,病毒和内毒素,可以激活免疫反应,从而促进炎性细胞因子短时间大量释放,并伴有大量的活性氧(ROS)[2]的产生,它直接损害心脏功能,最终导致患者的死亡。另外除了病原生物感染外,败血症或败血症性休克也是一种潜在的致命疾病,与强烈的全身性炎症反应有关,最终可能导致心肌严重的炎症异常。脂多糖(LPS),也称为内毒素[3],由细菌释放,促进促炎细胞因子,一氧化氮和类二十烷酸的分泌,导致脓毒性休克期间的中毒性心肌损伤。心肌炎心细胞凋亡发生发展[4]。
微小RNA(miRNA)是由18 ~25 个核苷酸组成的大型非编码小RNA 家族。miRNA 在转录后调节其靶基因的降解和翻译[5]。miRNA 参与炎症反应和自身免疫性疾病的发展,如类风湿性关节炎和多发性硬化[6]。越来越多的证据表明miRNA 参与心肌细胞凋亡的调控过程。例如,miR-101 和miR-29b均靶向Mcl-1 以防止细胞凋亡,而miR-499a 通过抑制组蛋白脱乙酰酶8 的表达而损害肌细胞存活。此外,miR-34a 通过下调SIRT1 的表达和诱导 p53 途径诱导细胞凋亡,而miR-16 通过靶标参与细胞活力和凋亡。细胞凋亡是一种程序性细胞死亡,是体内组织更新和器官发育的重要生物学过程,也可诱导病理生理变化[7]。例如,心肌细胞凋亡可能导致不可逆的心肌损伤,因此参与多种病理过程,从而导致各种心血管疾病,包括心肌炎、心力衰竭(HF)等。因此,人们普遍认为抑制细胞凋亡可以改善心脏组织中的病理变化并有益于改善各种心脏疾病中的心脏功能[8]。
最近的研究显示[9],与对照心脏相比,大鼠心脏衰竭模型中miRNA 的表达具有差异性,发现上调的 miRNA 有 79 种,28 种 miRNA 下调。然而在那些上调的miRNA 中,发现与对照心脏相比,衰竭心脏中miR-199a 的表达高 3.4 倍。这些发现都表明miRNA 在心脏稳态中的潜在作用。最近的研究表明miR-199a 可以通过抑制自噬和促进细胞凋亡来破坏血管内皮细胞,从而加重原发性高血压。另外近来的研究发现抑制miR-199a 对于心肌纤维化具有重要的作用。心肌炎是临床的发病临床较高的疾病,因此我们把焦点集中在miR-199a 对于心肌炎的作用上。这对于研究miR-199a 对于心肌炎损伤的作用具有重要的意义。
1 材料和方法
1.1 实验动物
60 只成年的SPF 级Wistar 大鼠购买于首都医科大学实验动物科学部[SCXK(京) 2018-0019],月龄为 2~3 个月,性别不限,体重 240~260 g。无菌手术在河南科技大学第一附属医院实验设施进行[SYXK(豫)2017-0011]。实验获得我院伦理委员会审批([201810]号),并按实验动物使用的3R 原则予以人道的关怀。
1.2 主要试剂与仪器
miR-199a minic(模拟物)、miR-199a inhibitor(抑制剂)购自于锐博;凋亡检测试剂盒(货号:FA101)试剂盒购自于全式金生物;p65 抗体(货号:8242T)、P65 抗 体 (货 号:3033T)、iκBa (货 号:4814T)、p-iκBa(货号:2859T)抗体,以上抗体均购自于 CST 公司。酶标仪购自于 INFINITE;Western blot 实验转膜仪、电泳仪等购自于伯乐;Evodiamine购自于Selleck。
1.3 实验方法
1.3.1 大鼠心肌细胞的提取
无菌条件下使用无菌剪取大鼠心室肌组织,将提取的组织放于D-Hank 液中,并清洗去除血细胞;随后将组织剪成小块,转移至50 mL 离心管中,静止2 min 弃上清,加入10 mL 0.1%的胰酶,用无菌吸管将组织吹匀并放入37℃水浴锅中。水浴加热消化15 min,每 3 min 混匀 1 次。弃上清。重复 2 次;随后按上述重新加入0.1%胰酶10 mL,消化15 min,后加入含10% FBS 的培养基终止消化并收集上清液。重复上述消化、收集细胞、终止消化等步骤5次,至组织变成白色。将收集的上清液按800 r/min离心,并用含10% FBS 的DMEM 培养基重悬。随后培养并进行鉴定,发现纯度>98%。
1.3.2 实验分组
将大鼠原代心肌细胞分为对照组(NC)、LPS组、LPS+miR-199a mimic 组、LPS+miR-199a inhibitor组;LPS 诱导剂量为 10 μg/mL。
1.3.3 CCK8 法检测各组心肌细胞的活力水平
实验分组同前,每孔铺8000 个细胞,每组各8个副孔。细胞贴壁后进行模拟物及抑制剂干预,干预8 h 后进行LPS 处理,24 h 后进行 CCK8 实验检测大鼠原代心肌活力水平。
1.3.4 Western blot 法检测各组蛋白表达
将大鼠原代心肌细胞分为对照组(NC)、LPS组、LPS+miR-199a mimic 组、LPS+miR-199a inhibitor组。各组每皿铺8×105个细胞。待细胞贴壁后进行miR-199a 模拟物或抑制剂处理。干预处理8 h 后进行LPS 干预,24 h 后裂解液进行裂解,提取蛋白。随后进行蛋白凝胶电泳及显像分析。
1.3.5 RT-qPCR 法检测各组大鼠原代心肌细胞中miR-199a 情况
运用TRIzol 法提取大鼠原代心肌细胞中的总RNA,随后进行逆转录。逆转录体系为20 μL:1 μL总 RNA,4 μL 5×Reaction Mix,2 μL 10×Super Script Enzyme Mix,20 μL 双蒸水,轻轻混匀后离心。随后在37℃下60 min,95℃下5 min 下进行逆转录。逆转录完成后,随后进行qPCR,总体系50 μL:1 μL cDNA,2 μL Mix,上下游引物各 2 μL,ddH2O 补足至50 μL;反应条件:95℃,预变性 5 min;95℃,变性30 s,67℃,延伸15 s,共40 个循环。基因的相对表达量用2-△△CT法进行计算。
引物序列如下:
β-actin 上 游 序 列: 5’-CATTGCTGACAGGAT GCAGAAGG-3’; 下 游 序 列: 5’-TGCTGGAAGGT GGACAGTGAGG-3’;miR-199a 上游序列:5’-CCAG TGTTCAGACTACC-3’,下 游 序 列: 5 ’-GAACA TGTCTGCGTATCTC-3’;
1.3.6 流式细胞仪测定各组大鼠原代心肌细胞凋亡差异
实验分组同前。1、细胞铺板,6 孔板每孔铺2×105个细胞。贴壁后进行相应处理。2、LPS 处理24 h后进行凋亡检测,收集培养液上清,终止消化后,3000 r/min 离心。随后弃上清,用 100 μL AV Buffer 重悬细胞,重悬后按 1:10 的比例加入Annexin V-FITC 进行常温孵育20 min,上机前加入碘化丙啶进行染色。
1.3.7 ELISA 法测各组中 TNF-α 和 IL-1β 含量
根据ELISA 试剂盒(Invitrogen,CA,USA)操作指导,取大鼠原代心肌细胞上清液检测 TNF-α 和IL-1β 的水平。用酶标仪(BioTekPowerWave XS,Winooski,VT,USA)在450 nm 的波长下测定标准品和样品。每个实验重复3 次。
1.3.8 抑制 NF-κB 通路研究 miR-199a 对于 LPS诱导后心肌细胞的影响。
实验分为对照组(con)、LPS 组、LPS+miR-199a mimic 组、LPS+miR-199a mimic+Evodiamine 组,利用NFκB 信号通路抑制剂 Evodiamine,研究其对于miR-199a 处理后细胞的作用。
1.4 统计学方法
统计学分析结果使用SPSS 18.0 软件分析。计量资料以平均数±标准差(±s)表示,两组间比较采用t检验;多组间比较采用单因素方差分析。P<0.05为差异有统计学意义。
2 结果
2.1 miR-199a 抑制了心肌炎时心肌细胞的活力
如图1 所示,为了研究miR-199a 对于心肌炎时心肌细胞的活力作用。与对照组相比,LPS 抑制了大鼠原代心肌细胞的活力(P<0.01),然而LPS诱导后经模拟物处理后,大鼠原代心肌活力受抑制更加严重(P<0.05)。表明 miR-199a 在心肌炎中可能加重了心肌细胞的损伤,同时,miR-199a 抑制剂组表明,与模拟物相比,抑制miR-199a 后发现LPS 诱导的大鼠原代心肌细胞活力受抑制降低(P<0.01)。
2.2 miR-199a 促进了心肌炎时心肌细胞炎症因子的释放
ELISA 结果如图2 所示,与 NC 组相比,LPS 诱导了 TNF-α 及 IL-1β 的释放(P<0.05)。与 LPS 组相比,miR-199a 模拟物组加重了 TNF-α(P<0.01)及 IL-1β(P<0.05)的释放。然而,经过 miR-199a 抑制剂处理后,TNF-α(P<0.01)及 IL-1β(P<0.05)的释放减少。
2.3 miR-199a 加重了心肌炎时心肌细胞的凋亡
为了研究心肌炎时miR-199a 在心肌细胞中的凋亡效应,如图3 所示,流式凋亡结果表明,与对照组相比,LPS 诱导了大鼠原代心肌细胞的凋亡(P<0.01),应用 50 nmol/L 的 miR-199a 模拟物后,与LPS 组相比,大鼠原代心肌细胞凋亡比例上调(P<0.05)。与模拟物相比,miR-199a 抑制剂组,大鼠原代心肌细胞凋亡明显降低(P<0.01)。表明miR-199a 可能通过影响凋亡来加重心肌炎的损伤。

图1 miR-199a 抑制了心肌炎时心肌细胞的活力Figure 1 miR-199a inhibits cardiomyocyte proliferation during myocarditis

图2 miR-199a 促进了心肌炎时心肌细胞炎症因子的释放Figure 2 miR-199a promotes the release of inflammatory cytokines in myocarditis
2.4 miR-199a 上调了心肌炎时心肌细胞凋亡相关蛋白的表达
如图4 所示与对照组相比,LPS 诱导组凋亡相关蛋白变化。LPS 诱导后,caspsse3、bax 表达上调,bcl2 表达下调。表明LPS 诱导了心肌细胞的凋亡。然而,模拟物组加重了LPS 诱导的凋亡蛋白的变化。与此同时,抑制miR-199a 后发现,与模拟物组相比,凋亡蛋白 caspsse3、bax 表达下调,bcl2 表达上调。这表明miR-199a 加重了心肌细胞的的损伤。
2.5 miRNA-199a 加重了 LPS 诱导的 NF-κB 通路的激活
如图5 所示,与对照组相比,LPS 诱导了大鼠原代心肌细胞NF-κB 通路的激活,具体表现为p-iκBa与p-p65 表达的上调。但与LPS 组相比,在模拟物处理后,p-p65 和 p-iκBa 上调更加明显。与此同时,阻断microRNA 表达后,与模拟物组相比,p-p65和p-iκBa 表达下调。这些发现表明用模拟物处理可以加重体内LPS 诱导的心肌炎中NF-κB 信号传导途径的激活。
2.6 抑制 NF-κB 通路缓解 miR-199a 对于 LPS 诱导的细胞的损伤
如图6 所 示,利 用 NF-κB 通 路 抑 制 剂Evodiamine,发现与模拟物组相比,应用Evodiamine干预后,大鼠原代心肌细胞活力下降(P<0.01),这进一步说明,miR-199a 可加重心肌炎时心肌细胞的损伤,这种损伤主要通过 NF-κB 信号通路来实现的。

图3 miR-199a 加重了心肌炎时心肌细胞的凋亡Figure 3 miR-199a aggravates cardiomyocyte apoptosis in myocarditis

图4 miR-199a 上调了心肌炎时心肌细胞凋亡相关蛋白的表达Figure 4 miR-199a up-regulates the expression of cardiomyocyte apoptosis-related proteins in myocarditis

图5 miRNA-199a 加重了 LPS 诱导的 NFκB 通路的激活Figure 5 miR-199a aggravates the activation of LPS-induced NFκB pathway
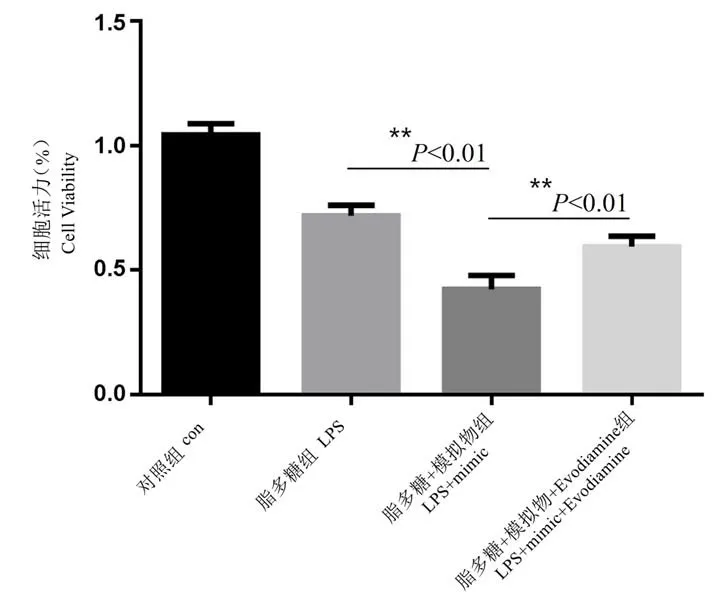
图6 抑制NF-κB 通路缓解miR-199a 对于LPS诱导的细胞的损伤Figure 6 Inhibition of NFκB pathway alleviates miR-199a damage to LPS-induced cells
3 讨论
目前关于心肌炎的发生发展机制尚未明确,但抑制心肌细胞凋亡来延缓心肌炎的进展已经成为共识[10]。NF-κB 是一核转录因子可被 LPS 诱导的TLR2 信号通路所激活。其由 Rel、P65、RelB、P50、P52 五个亚基构成[11]。在未激活的前提下,NF-κB与IκK(IκB 激酶复合体)相结合,因此处于失活的状态。因此 IκK 是激活 NF-κB 信号传导通路的主要环节。其激活分为经典途径与非经典途径两种方式。经典途径为,当刺激激活 IκK 后,IκB 蛋白被磷酸化,NF-κB 与 IκK 分离,失去抑制作用的 NF-κB入核发挥生物学作用。在非经典途径中,P100 被降解为强活性的P52,其与RelB 相互作用入核发挥转录活性[12]。综上NF-κB 信号通路促进参与调节细胞转化,细胞凋亡和对多种刺激的反应的各种生物学过程,例如应激,细胞因子和自由基,其在调节免疫应答中起关键作用。然而,过度激活的NF-κB 途径可能诱发持续和长期的系统性炎症,这是急性心肌病变的主要原因之一[13]。有研究表明通过激活Nrf2 / HO-1 途径抑制NF-κB 信号通路后可减轻糖尿病引起心肌的氧化应激和炎性渗出[14]。因此,阻断NF-κB 通路的不当激活,特别是阻止转录因子p65 的核转位,有助于阻断自身免疫系统的过度活化[15]。NF-κB 通路的激活对于心肌细胞中促炎细胞因子的产生是必需的。MicroRNA-199a(miR-199a)是一种新型基因调节因子,在炎症和损伤中扮演着重要作用。我们发现MicroRNA-199a 被抑制后可靶向SFRP5 来抑制心肌纤维化的进展[16]。其前体基因位于染色体1 和19 上。经剪切后,它转化为成熟的miR-199a-5p 和 miR-199a-3p。目前,miR-199a-5p 已得到更广泛的研究,揭示它与肿瘤细胞的活力,侵袭,迁移和自噬密切相关
在本研究中,我们通过CCK8 实验发现miR-199a 加重了心肌炎时大鼠原代心肌细胞的损伤,同时我们发现这种损伤可能是通过凋亡相关通路来实现的,这一结果都得到了流式凋亡及Western blot的支持,随后我们注意到LPS 诱导的心肌细胞NF-κB 信号通路得激活。然而,在 miR-199a mimic 组处理后,发现miR-199a 促进了核转录因子的核转位。与 LPS 组相比,miR-199a 引起 IκB 与 p-iκBa 的失衡,进而导致 P65 的磷酸化进而导致 NF-κB 的激活。从而加重了心肌炎时的炎症反应,加速了心肌细胞的凋亡。应用Evodiamine 干预后,大鼠原代心肌细胞活力下降,因此miR-199a 可能在心肌炎中进一步激活NF-κB 信号通路来加重心肌细胞的损伤。
