青年男性颜面和四肢远端萎缩伴皮肤变硬革化15年
——成人早老症
2020-11-06邹克季朱敏洪道俊
邹克季 朱敏 洪道俊
1 临床资料
患者男,22岁,因“颜面和四肢远端萎缩伴皮肤变硬革化15年”于2019年5月9日就诊于我院。
患者自7岁左右开始逐渐出现颜面部,主要为双眼周围和双侧颞部,以及耳根部皮肤粗糙,眼角有放射状皮肤皱纹,伴随皮肤脱屑,局部轻度色素沉着。约数个月后发现四肢远端皮肤粗糙,逐渐发展为远端肢体变硬萎缩,向近心端发展,目前表现为肘部及膝部以远的肢体萎缩变细,局部轻度色素沉着和脱屑,且皮肤干燥无汗。症状持续进展,无缓解复发,无肢体无力,无感觉障碍,无发热,无眼干和口干,无皮疹,无肌肉关节肿痛,无肢体僵硬。曾在当地医院多次就诊,未予特殊治疗,因不影响日常生活,患者一直没有重视上述症状,近期因找女朋友,女方说“肢体像老人家一样”而开始积极就医。自发病以来,进食正常,睡眠正常,二便功能正常。
既往史:幼年发育正常,自11岁起生长发育减慢,身高低于同龄人,智力及体育成绩正常。11年前逐渐出现左眼视力减退,诊断为“近视”,未予进一步检查及治疗。2019年2月于当地医院查体,诊断为“甲状腺功能减退”,给予左甲状腺素钠12.5 μg qd治疗。否认冠心病,糖尿病病史。吸烟史10余年,10支/d,未戒烟,间断饮酒史。无特殊药物和毒物接触史。父母非近亲结婚,现身体健康,否认相关家族史。
体格检查:体温36.7℃,心率73次/min,呼吸17次/min,血压122 mmHg/80 mmHg,身高 165 cm,双眼周围、双侧颞部,及耳根部皮肤粗糙紧绷,眼角有放射状皮肤皱纹及散在小片状色素沉着(图1A)。双手和前臂(图1B)、及双足和膝关节以远(图1C)皮肤紧绷变薄和萎缩,掌指、肘、膝、趾跖关节伸面有角质增生,小腿下1/3以远伴随皮肤脱屑,双足扁平。心、肺、腹检查无异常。
神经系统专科检查:意识清楚,声音嘶哑,对答切题,记忆力、执行力、计算力、定向力、逻辑判断能力正常;左眼视力粗侧0.3,右眼视力正常,双眼球共轭居中,双侧瞳孔等大等圆,对光反射对称存在,闭目鼓腮正常,双侧听力粗侧正常,吞咽正常,伸舌居中,无舌肌萎缩及纤颤。四肢远端肌容积减少,肌张力适中,肌力5级。双侧针刺痛觉、音叉振动觉对称存在。双侧肱二头肌、肱三头肌、桡骨膜、膝腱及跟腱反射对等引出。双侧Babinski′s征阴性。双侧指鼻及跟膝胫试验稳准。颈软,Kernig′s征阴性。
辅助检查:血尿便常规、肝功能、肾功能、血脂均正常。游离 T3:4.26 pmol/L (正常值 3.8~6.0 pmol/L), 游离 T4:12.1 pmol/L(正常值 7.9~15.4 pmol/L),促甲状腺素:27.37 mIU/L(正常值 0.34~5.60 mIU/L),早 8 点 ACTH:15.27 ng/L(正常值 6~40 pg/mL),下午点 4 点 ACTH:16.82 ng/L(正常值 3~30 pg/mL),空腹血糖、胰岛素及C肽、催乳素、生长激素等内分泌激素未见明显异常。血沉、抗核抗体、抗双链DNA抗体、ANCA抗体、ENA抗体谱、甲状腺球蛋白抗体、过氧化物酶抗体等免疫相关检测均阴性。心电图提示窦性心律,窦房结内游走,II导p波高尖。胸片显示两肺未见活动性病变,脊柱轻度向右侧弯。甲状腺彩超:甲状腺体积偏小伴质地不均,甲状腺双侧叶结节(TI-RADS 3级)。腹部B超:脂肪肝,胆、胆管、胰、脾、肾声像图未见明显异常。四肢肌电图提示运动和感觉神经波幅、传导速度及潜伏期正常。静息状态针极肌电图提示左侧股四头肌、右侧肱二头肌、左侧胫前肌可见少量自发电位;右侧股四头肌、左侧三角肌、左侧外展小指肌、右侧第一骨间肌、右侧胫前肌未见自发电位。在肌肉轻收缩下双侧股四头肌、右侧胫前肌、左侧三角肌、右侧肱二头肌、左侧外展小指肌可见时限增宽、部分电压增高,左侧胫前肌、右侧第一骨间肌时限电压正常。头颅MRI未见异常。
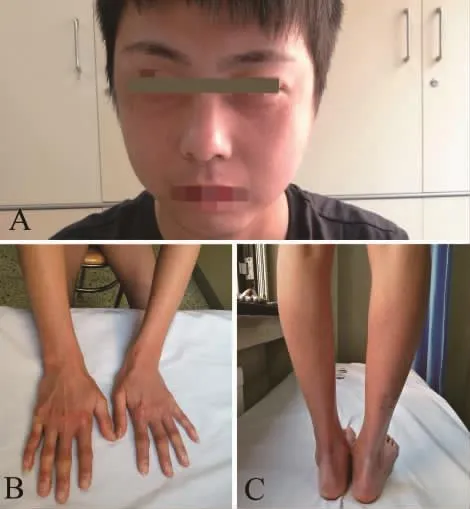
图1 患者颜面与四肢皮肤变化 患者双眼周围和颞部皮肤粗糙紧绷,眼角有放射状皮肤皱纹及散在小片状色素沉着(A)。双手和前臂(B)、双足和膝关节以远(C)皮肤紧绷变薄和萎缩,角质增生。
病理检查:经知情同意,对患者右下肢腓肠肌、腓肠神经、皮肤进行联合活检。皮肤组织HE染色光镜下显示皮肤角质层增厚,真皮层萎缩、皮肤附属器官(毛囊、汗腺)减少(图2A)。电镜下皮肤组织内的神经末梢部分大有髓神经纤维轴索空泡化(图2C),无髓神经纤维明显减少,出现胶原袋结构(图2D),无髓神经结构紊乱,个别出现细丝物质沉积。腓肠肌肌肉活检可见个别成小组分布的小角状萎缩肌纤维,个别靠近肌束衣、筋膜下的区域可见小圆状略萎缩肌纤维,部分肌纤维膜下线粒体略增多,可见小片状群组化改变。腓肠神经光镜有髓神经肌纤维的密度和结构基本正常(图3A和B),但无髓神经纤维数量明显减少(图3C)。腓肠神经电镜有髓神经纤维结构正常,无髓纤维减少(图 3D)。
基因检查:二代测序揭示了WRN基因上存在c.3139-1G>C和c.1960C>T复合杂合突变。家系验证结果显示此两个杂合突变分别来自于其父母,符合常染色体隐性遗传规律。突变位点c.3139-1G>C为剪切位点突变,对蛋白功能的影响可能较大,目前已有文献报道为致病突变(PubMed_ID:8602509)。根据HGMDpro数据库报道情况,突变位点c.1960C>T未见报道,突变位点c.1960C>T依据ACMG指南,其突变变异类型评级为Pathogenic(致病性突变),该突变为无义突变,导致蛋白翻译终止,对蛋白功能的影响可能较大。因此,根据临床表现,病理改变特点,以及基因突变结果,该患者诊断为成人早老症(Werner syndrome,WS)。
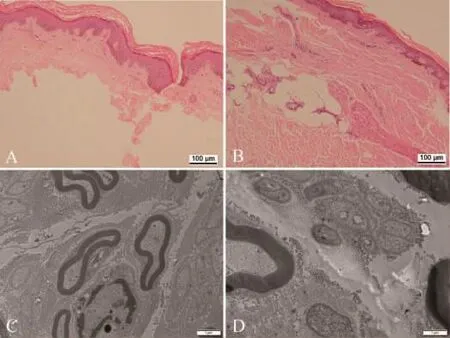
图2 皮肤组织HE染色 光镜下显示皮肤角质层增厚,表皮和真皮层萎缩、皮肤附属器官(毛囊、汗腺)减少(A,HE染色),而正常对照皮肤未见异常(B,HE染色)。电镜下皮肤组织内的神经末梢大有髓神经纤维轴索变性(C),无髓神经纤维明显减少,出现胶原袋结构(D)。
2 讨论
成人早老症是一种罕见的常染色体隐性遗传疾病,绝大多数患者在青春期前发育基本正常,进入青春期后开始加速衰老,临床特征与生理性衰老类似[1]。WS是由WRN基因发生功能缺失突变引起,该基因能够编码DNA解旋酶RecQ家族的WRN蛋白,其参与DNA复制、转录、修复、重组、端粒和着丝粒区异染色质维持等一系列细胞过程[2]。目前WS国际发病率约为1/100万~1000万,但在日本发病率高达1/10万,我国尚无相关流行病学报告[3]。
WS患者最常见的首发症状为缺乏青少年期的生长高峰,因而出现身材矮小和低体重,但因为不具有特异性,通常在病程回顾时才被意识到[4]。具有特征性的临床表现为患者20~30岁开始出现皮肤皱缩、皮下脂肪丢失、头发灰白和脱落等衰老相关特征[5]。本例患者在11岁时即出现生长发育较同龄人落后,一直没有得到重视,直到WS诊断明确后才被认为是该患者的一个临床症状。该患者在儿童期即出现颜面部的皮肤粗糙,进而发展为皱缩和脱屑,以及四肢远端皮肤的变硬变薄和脂肪萎缩,较经典的WS患者的皮肤损害出现早,提示个别WS患者的皮肤损害有可能作为首发症状出现。本例患者在就诊时上没有出现毛发脱落或者灰白改变,也间接导致了该患者临床的诊断难度,最新的诊断指南中特意提到在询问病史和查体时,要注意患者是否存在染发或植发的情况[6]。

图3 腓肠神经光镜有髓神经肌纤维的密度和结构基本正常 A:刚果红染色;B:神经丝蛋白免疫组化;C:半薄甲苯胺蓝染色显示大有髓神经纤维结构基本正常,但无髓神经纤维数量明显减少;D:腓肠神经电镜有髓神经纤维结构正常,无髓纤维减少。
WS患者随着年龄的增加,在40岁左右出现多系统的老化症状,主要包括:双眼白内障、声音嘶哑、2型糖尿病、性腺功能减退、骨质疏松、足跟部顽固性溃疡、跟腱骨化、动脉硬化、心脑血管事件、恶性肿瘤等[7]。肿瘤及心肌梗死是最主要的死亡原因,该病预后较差,平均寿命为54岁[8]。除了前述的皮肤异常和肢端脂肪萎缩之外,该例患者还存在扁平足、声音嘶哑、甲状腺功能低下等损害。按照2013年的诊断标准,临床方面达到了可疑的诊断标准,即2个核心症状(皮肤损害和声音嘶哑)+3个其他症状(发育落后、内分泌腺功能低下、骨骼异常)[6]。值得一提的是该患者在11岁即出现了单眼视力改变,而WS患者多伴有白内障导致的视力下降,遗憾的是患者拒绝行进一步的视力相关检查,而无法明确患者视力下降的原因。
皮肤皱缩和皮下脂肪丢失是WS患者的常见症状,其皮肤病理改变为表皮和真皮层萎缩,表皮脊消失[9-10]。该病例的皮肤病理改变也出现显著的表皮和真皮层的萎缩,但表皮脊尚有部分保留,可能与患者尚处于病程中早期相关。关于WS患者的神经活检病理改变仅有2例患者报道,均表现为个别的薄髓鞘纤维,而缺乏特异性的病理改变特点[11-12]。该例患者的神经活检显示无髓纤维和小纤维的显著丢失,而且在皮肤的末梢神经中也发现了类似的病理改变特点,提示患者存在小纤维神经病。此病理改变似乎和临床上患者肢端皮肤营养不良和汗腺的功能障碍相吻合,但两者是因果关系,还是伴随关系,以及是否具有普适价值尚需要进一步的观察和研究。此外,该病例提供了首例WS患者的肌肉活检病理观察,个别肌纤维内线粒体的异常聚集符合肌肉老化改变。
目前已知WS患者均由WRN蛋白的功能缺失突变(loss of function)导致,文献报道的WS致病突变通常为WRN基因的终止突变或者移码突变,导致WRN蛋白功能的丧失[13-14]。该例患者来自父源的c.1960C>T突变为终止码突变,导致WRN蛋白在654位氨基酸发生提前终止而发生蛋白截断。来自母源的c.3139-1G>C突变为剪切位点突变,理论上导致25号内含子的剪切异常,可能导致WRN的移码突变,进而导致WRN的截断型。因此,从该患者携带的两个复合杂合突变共分离现象分析,可以确定WRN基因发生的这两个突变是导致该患者致病的原因。WRN蛋白是一种多功能蛋白,主要负责细胞内遗传物质的复制和修复,当WRN蛋白功能缺失时,由于细胞内DNA不能维持稳态,导致细胞早衰或者癌变发生[15]。
WS患者主要需要和一组不典型早衰综合征鉴别,这其中包括不典型WS(atypical WS)由LMNA基因突变导致[16],哈钦森-吉尔福德早衰综合征 (Hutchinson-Gilford progeria syndrome,HGPS)由 LMNA 基因突变导致[17],Rothmund-Thomson综合征由 RECQ4基因突变导致[18],Bloom综合征由RECQ2基因突变导致[19]。这些患者表现为从儿童早期开始的衰老加速综合征,而缺少癌症风险增高、退行性关节病等WS症状。此外,WS也要注意和部分型早老症(如肢皮衰老、变性性早老症),以及其他原因导致的生长停滞相鉴别。
WS目前尚无有效的治疗方法。研究表明WRN功能异常将导致干细胞衰竭、线粒体功能障碍、自噬失衡等变化[20],因此多采用体内抑制GES细胞、维生素C、APM等抗氧化,雷帕霉素增强自噬等治疗手段。有研究团队提出了基因治疗策略,如应用法尼基化的抑制剂、反义寡核苷酸和 RNA干扰方法[21-22]。WS患者绝大部分死于肿瘤及心肌梗死,因此早期对心血管事件管理及肿瘤筛查十分重要。
3 点评
WS患者常表现为四肢远端的萎缩,因而可能被认为是周围神经病变或者远端型肌病而就诊于神经内科。WS患者肢体远端萎缩的本质是脂肪萎缩和皮肤老化,肌力和感觉相对正常。在本例患者中,我们通过系统的病理学技术观察到WS的小纤维神经存在明确的损害,可能与皮肤干燥和脂肪萎缩之间有一定的关系。然而两者之间的关系是因果关系,还是一种并存的病理现象,或是特殊病例的一种巧合,尚需要进一步的病例积累和研究。
转化医学(translational medicine)是将基础研究和临床研究连接起来的一种新的思维方式,早老症的研究就是转化医学研究的典范。从OTTO WERNER医生在1904年首次描述WS患者开始[23],科学家们对这种奇怪疾病的发病机制一直困惑不已,一直到1996年YU等[24]才成功克隆到了WRN基因。此后科学家对WRN蛋白的功能进行了深入的研究,发现WRN蛋白参与DNA复制、转录、修复、重组、端粒和着丝粒区异染色质维持等一系列保护细胞遗传物质稳态的生理过程[2]。在这个研究过程中,科学家后续还发现了LAMN、RECQ2、RECQ4等一些导致其他类似早老症的基因,这些基因共同的特点都参与细胞核内遗传物质稳态的维持。基于这些研究,科学家终于对细胞衰老和老化有了深层次的理解,也就是说衰老的研究很大程度上是因为临床上的发现,通过转化为基础研究,进而推动了该领域的发展[25]。事实上这也是临床遗传学家热衷于遗传病研究的一个重要原因,有时通过一个新基因的克隆,可能为生命的基础研究突然打开一扇窗,开辟一个新的领域。
