应用单核苷酸多态性微阵列技术对不明原因智力低下/发育迟缓患儿的遗传学分析*
2020-11-04熊卿圆岑锦明曾赤佳
熊卿圆, 岑锦明, 曾赤佳
1佛山市禅城区中心医院检验科(广东佛山 528000); 2佛山市第一人民医院心血管内科(广东佛山 528000)
婴幼儿不明原因的精神发育迟缓(mental retardation, MR),也称为智力落后(mental deficiency, MD),为小儿常见的一种发育障碍(developmental delay,DD),是中枢神经系统功能障碍的一种可变表现,在人群中发生率接近3%[1],可由遗传、环境、围生期缺氧窒息等引起[2],其中遗传因素约占40%。应用传统的G显带染色体核型分析检出异常最多的婴幼儿问题为:精神运动发育迟缓(MR/DD),孤独症谱系障碍(autism spectrum disorders,ASD),包括先天性心脏病、脑发育不良、视觉听觉异常等的先天发育异常(congenital anomalies,CA)[3-4],但G显带核型分析只能检出染色体5~10 Mb以上的异常,而荧光原位杂交(fluorescence in situ hybridization,FISH)技术只能检出特定的微小异常。只有低于6%的患者能通过传统染色体核型分析和FISH技术检测出异常[5]。染色体微阵列技术(chromosomal microarray analysis,CMA)又称为“分子核型分析”, 可检测染色体不平衡的拷贝数变异(copy number variant, CNV),尤其是对于检测染色体组微小缺失、重复等不平衡性重排具有突出优势。根据芯片设计与检测原理,CMA可分为基于微阵列的比较基因组杂交(array-based comparative genomic hybridization, aCGH)技术和单核苷酸多态性微阵列(single nucleotide polymorphism array, SNP array)技术[3]两大类,其中SNP array除可检出拷贝数变异外,还可检出单亲二倍体、杂合性缺失(LOH)、纯合状态(AOH)和三倍体。CNV可引起包括神经发育性疾病、天性心脏病等先天发育异常,而CMA是先天发育异常和神经发育性疾病诊断的一线检测技术,但在提高异常检出率的同时,也会检出许多临床意义不明确的变异(varians of unknown clinical significance,VOUS),所以,对结果的解读非常重要。本研究应用染色体核型分析与单核苷酸多态性微阵列技术对168例不明原因的精神运动发育迟缓患儿进行扫描与分析,探讨结果的解读,以评估其遗传病因,为进一步治疗及遗传咨询提供依据。
1 资料与方法
1.1 一般资料 2015年7月至2019年6月来我院小儿神经康复科门诊及儿童保健门诊就诊的不明原因MR/DD患儿168例,根据MR/DD诊断及筛查标准[6],排除已知明确病因的疾病,如有明确的染色体核型异常、遗传代谢性疾病、围生期异常所致的缺氧缺血性脑病等,所有病例先行G显带染色体核型分析,未见明显异常或检出平衡易位及未知来源片段的标本再采用微阵列技术进行分析。所有患儿均为临床病例,入选患者家属在入选时均被告知其研究方法及意义,做到知情同意,并得到了佛山市禅城区中心医院医学伦理委员会的批准。
1.2 样本采集及预处理 在受试者安静状态下采集肝素抗凝及乙二胺四乙酸(EDTA)抗凝外周血各2 mL。
1.3 G显带染色体核型分析 肝素抗凝外周血0.5 mL接种至5 mL含PHA的外周血淋巴细胞培养基中,置37℃,5% CO2的二氧化碳培养箱中培养68~72 h,加入100 μg/mL秋水仙素50 μL,作用12 min后收获,将培养基移入15 mL离心管中,离心后吸去上清液,加入8 mL 0.75%KCl溶液低渗,置37℃水浴25 min,然后加入甲醇与冰乙酸按3∶1比例配制的固定液2 mL,混匀离心后吸去上清液,加入固定液8 mL,固定30 min后离心吸去上清液,再加入8 mL固定液重复固定一次,离心后视细胞的多少余留少许上清液制成合适浓度的细胞悬液。用吸管将细胞悬液混匀,吸取细胞悬液自10~20 cm高度滴在一干净并预先冷却的玻片上,在烤片机上缓慢烘干。65℃烤片3 h至过夜,老化后的玻片使用胰酶消化,吉姆萨母液用0.1 mol/L pH=7.4磷酸缓冲液稀释,染色胰酶消化后的玻片,除去多余的染液,晾干,在油镜下观察。所有核型结果均按照《人类细胞遗传学国际命名体制(2016)》(《An International System for Human Cytogenetic Nomenclature 2016》)进行描述。
1.4 CMA检测 乙二胺四乙酸(EDTA)抗凝外周血送至广州金域检验中心或广东省妇女儿童保健院医学遗传中心,进行CMA检测,两机构均采用Affymetrix公司配套检测试剂盒及优化的标准操作流程,严格按照质控标准进行DNA提取、酶切、连接、PCR、PCR产物纯化、片段化、标记、杂交等操作,使用 CytoScanTM750K芯片进行全基因组范围扫描,Affymetrix Chromosome Analysis Suite Software;version 3.1.0.15进行分析。并使用OMIM (Online Mendelian Inheritance in Man)、DECIPHER (Database of Chromosomal Imbalances and Phenotypes using Ensembl Resources)、 PubMed等数据库对结果进行比对分析。
2 结果
2.1 染色体核型分析结果 168例患儿中,检出染色体异常14例(8.33%)。14例异常患儿中,5例(35.71%)患儿有不同程度的特殊面部特征(眼脸外上斜、宽眼距、低鼻梁、高前额、小头畸形等),6例(42.85%)伴有其他先天发育异常,如先天性心脏病、脑发育不良、视觉听觉异常、多指畸形,基始子宫,隐睾等。14例患儿核型分析结果及临床表现见表1。
2.2 CMA检测结果 168例患儿中,154例G显带染色体核型分析未见明显异常患儿及表1中1-5号患儿共159例患儿使用单核苷酸多态性微阵列技术进行了进一步扫描分析,26例(16.35%)检出临床相关变异,其中24例存在CNVs,2例为整条染色体或大片段纯合子。检出的CNVs中,微缺失16例,微重复8例。

表1 G显带染色体核型分析检出异常病例结果及临床表现
检出致病的CNVs为18例,具体结果见表2。18例致病的20个CNVs中,大部分(16/20)>1 Mb,15例为微缺失,5例为微重复,微缺失是微重复的3倍;11例12个CNVs<5 Mb,13例有对应的综合征。
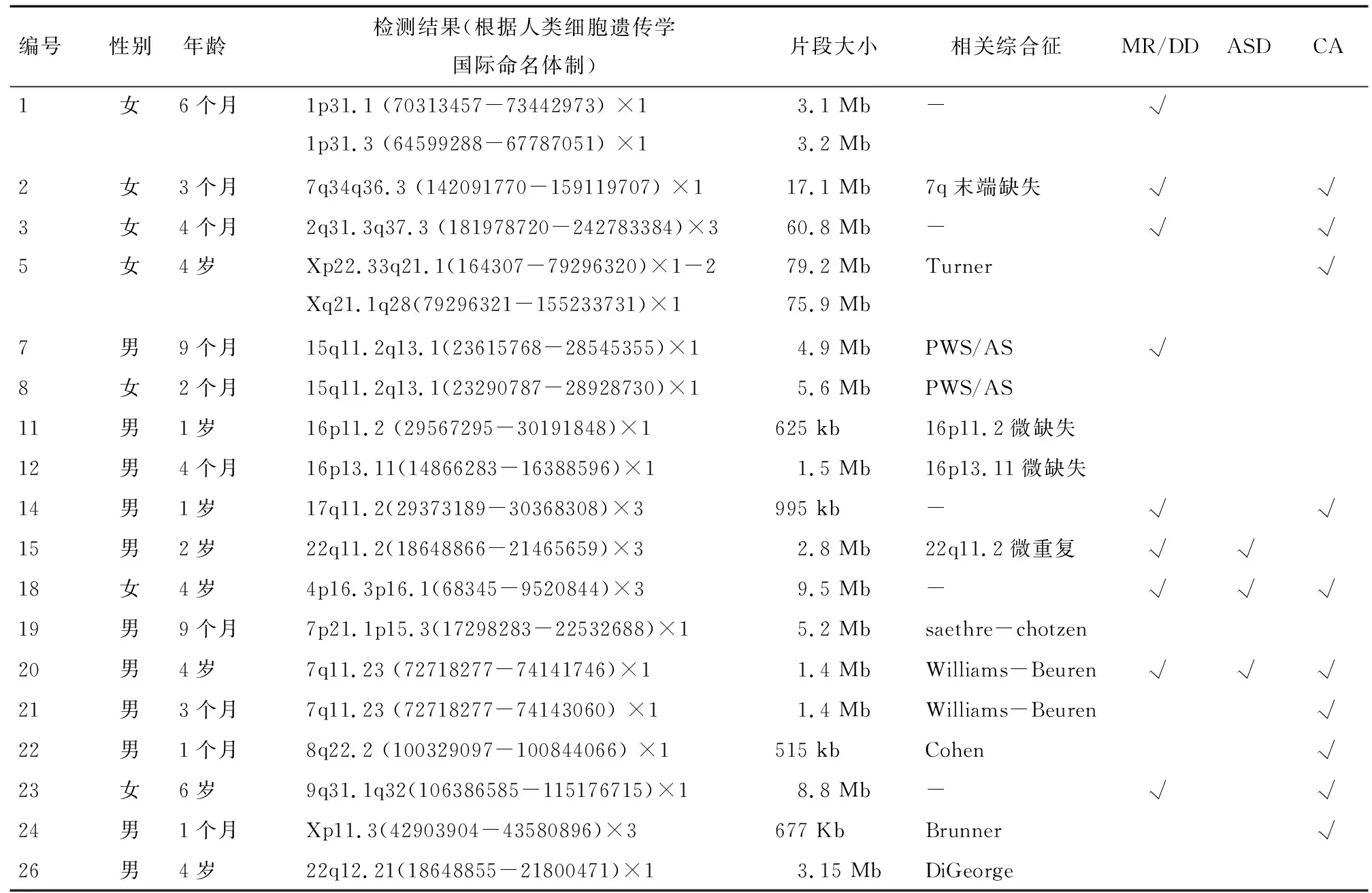
表2 SNP array 检出致病性CNVs结果
检出可能致病性变异4例,3例为微缺失,其中2例缺失片段在1~5 Mb之间,1例为纯合子,纯和片段>10 Mb。2例有对应的综合征。具体结果见表3。

表3 SNP array 检出可能致病性变异结果
检出临床意义不明的变异(VOUS)4例,具体结果见表4,其中3例为微重复,且重复片段均≤1Mb,1例为纯合子,纯和部分片段均>10 Mb。

表4 SNP array检出临床意义不明的变异结果
26例异常中,2例患儿同时有MR/DD、ASD合并CA,且均检出致病CNVs;8例患儿同时有MR/DD与CA,其中4例检出致病CNVs,2例检出可能致病CNVs,2例检出VOUS;1例患儿同时有MR/DD与ASD,且检出致病CNV;4例MR/DD患儿中,2例检出致病CNVs,检出可能致病CNVs和VOUS各1例; CA患儿则有3例检出致病CNVs,2例检出可能致病CNVs,1例检出VOUS。
检出异常最多的为7号染色体(4例),检出15、16、22、X号染色体异常均为2例;7q11.23与15q11.2均检出2例,最大异常片段为包括X整条长臂的79.2Mb缺失(5号病例)。
3 讨论
3.1 G显带染色体核型分析与SNP array异常检出率分析 168例患儿中,通过传统G显带染色体检出异常14例(8.33%),使用SNP array检测的159例患儿中,检出异常26例(16.35%)。通过染色体微阵列技术检出而未能用传统G显带染色体核型分析检出或检测为平衡易位的致病性CNV为16例(9.52%),可能是由于传统G显带染色体核型分析的分辨率为5~10 Mb,限制较大,与其相比,SNP array技术在检测小片段DNA拷贝数异常方面优势明显。
3.2 本研究SNP array结果分析及意义 检出的致病性CNVs的18个病例中,有13例有对应的已知综合征,其中与Angleman综合征(AS)和Prader-Willi综合征(PWS)相关、与Williams-Beuren综合征相关各2例,与7q末端缺失综合征、22q11.2 重复综合征、saethre-chotzen 综合征、Cohen综合征、Brunner 综合征、Turner综合征、16p11.2微缺失综合征、16p13.11微缺失综合征、DiGeorge综合征相关病例各1例。可见目前数据库所覆盖的CNVs已较全面,大部分CNVs已被临床所认识,SNP array检出结果可以很好地指导临床诊断。
检出的可能致病CNVs中,1例(4号病例)位于6p22-p24缺失致病区域内,但不包含关键基因ATXN1和JARMR2,6p22-p24缺失主要表现为智力低下、语言发育迟缓、注意力缺陷等行为异常、特殊面容、多发畸形等[7],但数据库内未见与本病例缺失片段完全一致的病例报道。1例(17号病例)包含重要功能基因ERBB4,一文献报道ERBB4基因发生缺失可引起智力障碍,语言发育迟缓,多动症等临床表现。Decipher数据库分别报道3个病例(271941,267738和252228)与本病例缺失大小相似,均涉及ERBB4基因,且两例为新生突变,临床表现为智力低下和语言发育迟缓。ERBB4基因功能缺失也可引起肌萎缩性脊髓侧索硬化症(Amyotrophic lateral sclero)[8-10]。1例(25号病例)为15号染色体大片段纯合子,包含了与AS/PWS相关基因[11]。1例(9号病例)包含4个基因,可能导致神经类精神类疾病发生,如智力低下、自闭症谱系疾病,癫痫和精神分裂症等,其他表现为肌张力低下和骨骼发育异常等[12]。
检出的临床意义不明VOUS中,1例(6号病例)包含30个基因,但未见该区域相关文献报道。1例(10号病例)包含22个基因,Decipher数据报道三例相似病例,一例报告为可疑良性变异(286187),两例报告为临床意义不明(294373,349736)。1例(13号病例)包含25个基因,与本片段相似重复病例相应报道记载的临床表现为智力低下、发育迟缓等(283139)。1例(16号病例)2号染色体发生大片段纯合子现象,可能为2号部分纯合,或整条2号染色体为单亲二倍体(uniparental disomy,UPD),无证据证明UPD(2)致病[13]。
结果发现,检出致病的CNVs中,微缺失是微重复的3倍,可能致病CNVs中也有3/4为微缺失,而VOUS中,3/4为微重复,可以推测,微缺失比微重复更易致病,而微重复为VOUS的可能性较大。结合临床数据和家族史信息能更好地分析CNV的遗传模式,但在实际应用中很难让双亲都接受检查,Iossifov等[14]认为,新发的CNV比遗传自亲代的更易致病, 如本研究26号病例,其父亲为表型正常的微重复(1q21.1q21.2(144034100-148662253)×3)携带者,其胞弟继承了父亲的基因型,暂未发现表型异常,而26号病例检测出新发致病CNVs,与DiGeroge综合征相关,该患儿表现为免疫力低下,出生至今多次因感染入院。另外,结果中检出2例大片段纯合子,可能为该染色体发生部分纯合,也可能为整条染色体发生单亲二倍体(UPD),两种均可能导致相应区域内隐性遗传病的发病风险增加。但由于CMA只能检测同源性UPD,而不能检测异源性UPD[13],所以更需要双亲进行检测,比对结果,以明确该染色体是部分纯合还是整条染色体发生单亲二倍体。所以,尽可能的完善家族史CMA信息对检出CNVs性质的判定非常有意义,也可更好地对患儿进行诊断及相应治疗。
包含多个基因或已知致病基因的CNV比只包含少量基因或所包含的为功能未知基因的CNV更易致病,可能是由于基因的剂量敏感度所致,由此可推论,大片段CNVs可能比小片段CNVs更易引起明显的临床症状。缺失可能会引起单倍剂量不足,一些小片段的微缺失也会引起相应的致病综合征,如8q22.2缺失515kb,涉及基因VPS13B,与Cohen综合征相关(22号病例),该综合征呈常染色体隐性遗传,其典型临床症状包括智力低下,产后小头畸形,面容异常(毛发浓密,发际线低,眼睑下垂,烧杯样鼻,高腭弓等),色素视网膜病变,近视,间断性中性粒细胞减少等[15]。另外一些小的缺失也可能导致患病,如15q11.2缺失312kb,包含TUBGCP5、CYFIP1、NIPA2、NIPA1 基因[16-17],可能导致精神类疾病发生,如智力低下、自闭症谱系疾病,癫痫和精神分裂症等,也可表现为肌张力低下和骨骼发育异常等[12]。而许多正常病例中检出大片段重复却不致病,甚至在正常人身上也可检出的情况另重复的机制变得很难解释,特别是若在相关基因调控区域或与临床相关的基因上则更加复杂。
根据患儿的表型结果分类显示,同时存在MR/DD与CA的患儿与检出的致病CNVs最多(4例),其次是CA(3例),再次是MR/DD、ASD合并CA患儿与MR/DD患儿(均为2例),最少是MR/DD合并ASD患儿(1例),可见先天异常与精神运动发育迟缓同时出现是染色体可能存在异常的一个重要的表现。结果中3例存在ASD患儿均检出致病性CNVs,Iossifov等[14]表明,ASD患者更有可能携带新发且罕见的CNVs和基因水平的突变。Schaefer等[18]报道CMA可以在约10%的ASD病例中检出有临床意义的CNV,全基因组测序和全外显子测序可能可以将ASD中阳性的检出率提高10%~15%[19],而CMA联合WES可以诊断约20%的ASD[20]。因此,将来可能会逐渐将测序作为主要的检测手段。
现在越来越多的实验室倾向于使用分辨率更高的CMA平台,预计将发现更多的致病CNVs,而这也将同时导致VOUS的数量增加。所以除了要注重基因型-表型的相关性以外,还可依据CNVs的大小和类型、遗传模式、基因型与表型之间的关系等来更好地解释单核苷酸多态性微阵列技术所检测到的CNVs结果,以更好地分析在整个基因组中的存在复杂相互作用的CNVs,为更多的不明原因MR/DD患儿提供明确的病因诊断,对深入研究MR/DD病因机制、患儿的预后及遗传咨询有重要的意义。
