Sam68基因对子宫内膜癌细胞增殖的影响*
2020-11-04梁常艳杨越波李小毛
梁常艳, 杨越波, 李小毛
中山大学附属第三医院妇科(广东广州 510630)
近20年来子宫内膜癌发生率在世界范围内呈上升趋势,为最常见的妇科肿瘤之一。随着人均期望寿命的延长,人口老龄化程度将加剧,子宫内膜癌高发人群的数量将逐年增长[1]。因此,子宫内膜癌的防治将成为我国肿瘤研究的重点领域之一。研究子宫内膜癌发生发展的分子机制并寻找新的治疗靶点, 是当前子宫内膜癌研究的热点。Sam68 (Src-associated in mitosis 68 kD) 作为信号转导与RNA激活(signal transduction and activation of RNA,STAR)家族成员之一,具有联系信号转导通路和RNA剪接的重要生物学功能。Sam68可能通过其SH2、SH3和(或)WW 结构域,与一系列信号转导分子,包括Src、ERK、PI3K、FBP21和FBP30等相互作用,从而发挥类似接头蛋白的作用,参与细胞增殖、生长和分化过程[2-3]。近年来研究发现, Sam68在宫颈癌、乳腺癌、卵巢癌、前列腺癌、鼻咽癌等实体肿瘤组织中较正常组织表达异常升高,与肿瘤生长、凋亡、侵袭转移等密切相关[4]。但是Sam68在子宫内膜癌中的具体作用目前尚未明确。2017年1月至2019年1月,本课题选择不同分化水平的子宫内膜癌细胞株(高分化Ishikawa,中分化HEC-1A,低分化AN3CA)作为研究对象,通过建立稳定干扰和过表达Sam68基因的子宫内膜癌细胞株,检测子宫内膜癌细胞增殖活力的改变,为进一步阐明Sam68在子宫内膜癌发生发展中的作用及其机制奠定实验基础。
1 材料与方法
1.1 细胞培养 人子宫内膜癌细胞株HEC-1A、 Ishikawa、RL952、JEC、AN3CA 购自美国American Type Culture Collection(ATCC)。将细胞接种在含有 10% 胎牛血清(HyClone, Logan, UT, USA)的DMEM 培养液(Invitrogen, Carlsbad, CA, USA),置于 37℃、5% CO2的培养箱中培养。待细胞生长到约80%,加入胰蛋白酶消化、传代,取对数生长期细胞进行后续实验。
1.2 实时定量 PCR和蛋白质印迹法分析 收集对数生长期的内膜癌细胞,使用Trizol试剂(Invitrogen)提取总RNA,逆转录成 cDNA(实验步骤参阅RNA反转录试剂盒Promega A3500 说明书),进行实时定量PCR 反应,同时以 β-actin 作为内参,检测Sam68 mRNA 水平的表达。Sam68 上游引物 5′-ATGAAGCTTATGGCCAGGAC-3′,下游引物 5′-CAGAAGCCAGAATGCAGAGT-3′;内参β-actin上游引物 5′-GAAGGTGAAGGTCGGAGC-3′,下游引物 5′-GAAGATGGTGATGGGATTTC-3′。实时定量PCR循环参数:95℃ 30 s,95℃ 5 s,60℃ 34 s,共 40 个循环。采用 2-ΔΔCt半定量法算出Sam68 mRNA与阴性对照组的相对表达水平。同时提取细胞总蛋白, BCA 法测定蛋白浓度,调整样本为浓度1 μg/μL,100℃ 水浴5 min 使蛋白变性,取 20 μg 蛋白进行 10% SDS-PAGE 凝胶电泳,转移至PVDF膜(Milipore,美国),用含 5% 脱脂牛奶封闭 1 h ;加入兔抗人 Sam68 单克隆抗体(体积稀释比为 1∶2 000)及兔抗人 β-actin 单克隆抗体(体积稀释比为1∶3 000),4℃反应过夜,TBS漂洗后分别孵育二抗。加入ECL 试剂,胶片曝光、显影和定影。图像分析系统扫描其吸光度值,进行定量分析。
1.3 子宫内膜癌细胞转染siRNA及过表达Sam68 根据Genbank中人Sam68基因序列,两个人类siRNA序列被克隆到pSuper-retro-puro质粒中产生分别pSuper-retro-Sam68-RNAi,序列为RNAi# 1: ggaccacaagaatACAATC和RNAi#2: GCATCCAGAGGATACCTTTGC(Invitrogen公司合成)。慢病毒转染方法参照Turbo FectTMsiRNA Transfection Reagent (Thermo,美国)试剂说明书。转染HEC-1A和RL952细胞株,Q-PCR和Western blot检测转染及沉默效果,具体按照1.2方法进行。从Ishikawa细胞中提取RNA并逆转录制备cDNA模板,扩增Sam68基因并将扩增片段克隆至真核表达载体pcDNA-3送测序进一步验证。利用Lipofecamine3000(Invitrogen,美国)转染pcDNA-3- Sam68至Ishikawa细胞,同时将pcDNA-3转染作为空白对照。Q-PCR和Western blot检测转染及过表达效果,具体按照1.2方法进行。
1.4 MTT法 将转染后的子宫内膜癌细胞按照104/孔的密度接种于96孔板,继续培养24、48、72、96、120 h后进行细胞活力检测。去除培养液,加入预先配置的MTT试剂,于恒温培养箱中静置30 min,取出细胞培养板置于酶标仪中读取490处吸光度OD值,计算细胞的增殖能力。同时设置调零孔、对照孔,每组设定3复孔。
1.5 平板克隆实验 消化收集各组对数生长期细胞,调整细胞悬液浓度至250个/mL,接种6孔板,每孔2 mL,即500细胞/孔。置于37℃、饱和湿度的5% CO2培养箱中培养10~14 d,观察克隆出现日期及生长情况,发现克隆集落即停止培养。1×PBS洗3次,加入4%多聚甲醛1 mL/孔固定10 min,加1 mL/孔苏木素染色10 min,用自来水轻轻冲洗干净。计数每个孔的克隆数并拍照。克隆形成率(%)=(克隆形成数/接种细胞数)×100%。
1.6 软琼脂(soft agar)克隆形成实验 软琼脂培养法原理是利用癌细胞接触抑制效应的丧失,能在悬浮状态下很好地成簇生长,检测肿瘤细胞永生化增殖能力。具体步骤参考相关文献[5]:在微波炉中溶解已消毒的1.2%琼脂,并将其与取1 mL 2×DMEM按照1∶1比例混合, 6孔板中每孔迅速加入1.5 mL混合液,每孔3 mL,待其自然冷却,此作为下层胶铺板;0.25%胰酶消化对数生长期的细胞,离心,将细胞悬浮于2× DMEM培养液中,计数后,使细胞浓度调整至为4×104·mL-1。 按照1∶1比例混合0.7%琼脂和2×DMEM培养液,充分混匀,加2 mL 混合后的含0.33%的1×DMEM到6孔板中的每个孔中,每孔含细胞数5×104。每个实验组重复3个样本,待上层琼脂凝固后, 置于5%CO2、37℃培养14 d。间隔2 d补加200 μL 1×DMEM到上层胶上,以防水分蒸发干燥。将平皿放置在倒置显微镜下,在镜下随机选择10个视野,计算视野中大于50个克隆数和所有克隆数,克隆形成率=大于50个克隆数/所有克隆数×100%。每孔加入1 mL的0.005%结晶紫染色1 h以上,镜下拍照。
1.7 BrdU细胞增殖实验 将状态良好的对数生长期细胞按照1×105/孔密度接种于24孔板中,待细胞贴壁后换无血清培养基饥饿细胞24 h,细胞密度达60%左右时每孔加入10 μL的BrdU工作液孵育60 min,每个实验组重复3孔。弃去培养基后PBS洗3次,4%多聚甲醛固定60 s,弃去固定液后PBS再次缓洗3次,加入0.1%曲拉通溶液破膜作用20 min,PBS洗后加入一抗anti-BrdU,室温孵育1 h,弃去一抗PBS清洗3次,加入二抗罗丹明试剂(注意避光)室温作用1 h,PBS洗清洗3次后加入DAPI对总体细胞进行核染30 min,PBS清洗、晾干、封片,荧光显微镜下观察细胞增殖情况。
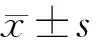
2 结果
2.1 人子宫内膜癌细胞中Sam68 mRNA及蛋白较正常子宫内膜组织表达升高 选取了5株不同分化的人子宫内膜癌细胞系(HEC-1A, Ishikawa, RL952, JEC和AN3CA)及人正常子宫内膜组织进行检测。实时荧光定量 PCR 法检测结果如图1-A显示,与人正常子宫内膜组织相比,人子宫内膜癌细胞系中Sam68 mRNA的表达均出现明显上调(图1-A)。蛋白质印迹法检测5株不同分化的人子宫内膜癌细胞系Sam68蛋白含量,结果提示,人子宫内膜癌细胞中Sam68蛋白的表达水平明显高于对照组(P值均<0.05)(图1-B)。后续实验中我们选用 Sam68 基因高表达的RL952(低分化)、HEC-1A(中分化)细胞进行RNAi沉默,用Sam68基因表达最低Ishikawa(高分化)构建过表达模型。

注:A:mRNA;B:蛋白子宫内膜癌细胞系中Sam68 mRNA和蛋白水平均高于正常子宫内膜组织,β-actin 作为内参(n=3)。*P<0.05,**P<0.001图1 实时荧光定量 PCR和蛋白质印迹法检测Sam68 mRNA和蛋白水平
2.2 成功构建Sam68 mRNA干扰及过表达子宫内膜癌细胞株模型 为了得到Sam68稳定敲减的细胞克隆,本实验采用shRNA介导的RNAi干扰技术下调Sam68的表达,采用实时荧光定量检测干扰效率。如图2-A所示,Sam68 RNAi#1和Sam68RNAi#2组敲减效率均大于80%,与阴性对照组相比,RL952和HEC-1A细胞可见Sam68 mRNA 表达显著降低(P<0.05)。进一步采用Western blot 实验验证在蛋白水平干扰载体对Sam68沉默的效率,与阴性对照组相比,已转染干扰病毒的RL952和HEC-1A细胞中,Sam68条带明显减弱,表明稳定转染干扰载体后,Sam68蛋白表达水平明显下调。以上结果表明,我们设计的两个人类siRNA序列均能成功有效敲除Sam68基因,成功建立了干扰Sam68的稳定细胞株。将pc DNA-3和 pc DNA-3-Sam68 质粒分别转染至Ishikawa 细胞 48 h 后,应用实时荧光定量 PCR 和蛋白质印迹法,检测转染的 Ishikawa细胞中 Sam68 mRNA 和蛋白的表达情况,如图 2-B显示,实验组pc DNA-3-Sam68 组细胞中 Sam68 mRNA 和蛋白的表达水平明显高于 pc DNA-3 组(P<0.05)。这一结果表明,pc DNA-3-Sam68重组质粒成功转染Ishikawa细胞后能够促进细胞 Sam68 蛋白的过表达。
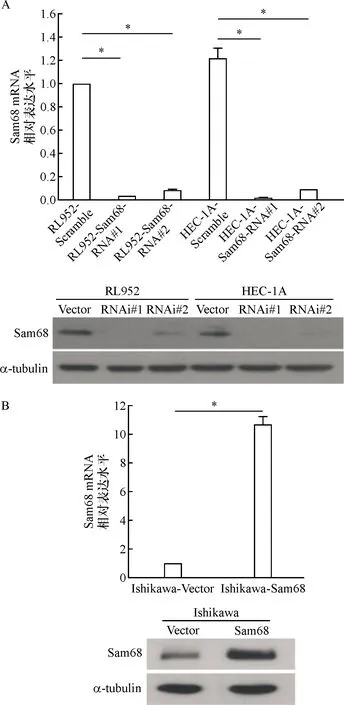
注:*P<0.05图2 实时荧光定量 PCR和蛋白质印迹法检测子宫内膜癌细胞中Sam68的基因和蛋白水平的敲减(A)与过表达效率(B)
2.3 敲除Sam68基因可抑制子宫内膜癌细胞增殖 HEC-1A和RL952细胞分别转染pSuper-retro-Sam68- RNAi#1和Sam68RNAi#2 和pSuper-retro-puro质粒(scramble组),对每组观测时间的吸光度值进行统计,分析显示Sam68- RNAi#1和Sam68RNAi#2干扰组活细胞数量明显减少,细胞生长速度变慢,且随着时间延长这种抑制作用更明显,与对照组比较,差异有统计学意义(P<0.05),见图3,提示下调Sam68基因可明显抑制子宫内膜癌细胞生长速度。平板克隆实验显示,转染RNAi组的细胞克隆形成率显著低于scramble组(P<0.001),见图4-A。图4-B软琼脂实验显示转染RNAi组的细胞形成直径>0.1 mm的集落数目明显低于scramble组(P<0.001)。BrdU细胞增殖实验是通过测定肿瘤细胞周期中S期细胞比例来准确反映肿瘤细胞的增殖情况。本实验通过荧光显微镜下拍照并统计数据显示,人子宫内膜癌HEC-1A和RL952转染Sam68RNAi质粒后,细胞处于S期(DNA分子中掺入BrdU细胞,显示红色)的比例明显低于scramble组(P<0.05),见图4-C。上述结果均说明Sam68 RNAi对人子宫内膜癌细胞的增殖能力抑制作用显著。
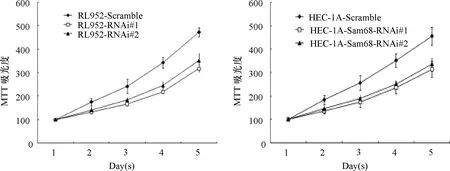
图3 MTT法检测敲除Sam68基因子宫内膜癌细胞增殖情况

注:A:平板克隆;B:软琼脂克隆形成; C:BrdU细胞增殖实验提示沉默Sam68 mRNA后子宫内膜癌细胞RL952和HEC-1A增殖受到明显抑制,*P<0.05图4 Sam68基因子宫内膜癌细胞增殖情况
2.4 过表达Sam68基因可促进子宫内膜癌细胞增殖 将pc DNA-3和 pc DNA-3-Sam68 质粒分别转染至Ishikawa 细胞 48 h 后,将细胞接种于96孔板,继续培养5 d,应用MTT检测各组细胞增殖情况,显示pc DNA-3-Sam68组Ishikawa 细胞在48、72和96 h这3个时间点的OD值均明显高于pc DNA-3组(P<0.05),见图5-C,这提示过表达Sam68基因可明显促进子宫内膜癌细胞生长。平板克隆实验显示转染pc DNA-3-Sam68组的细胞克隆形成率明显高于scramble组(P<0.001),见图5-A。图5-B软琼脂实验显示转染pc DNA-3-Sam68组的细胞形成直径>0.1 mm的集落数目明显高于scramble组(P<0.001)。BrdU细胞增殖实验显示,人子宫内膜癌Ishikawa转染pc DNA-3-Sam68质粒后,细胞处于S期的比例明显高于scramble组(P<0.05),见图5-D。上述结果均说明过表达Sam68基因可促进人子宫内膜癌细胞的增殖。

注:A:平板克隆;B:软琼脂克隆形成;C:MTT;D1、D2:BrdU细胞增殖实验提示过表达Sam68可促进子宫内膜癌细胞Ishikawa增殖,*P<0.05图5 过表达Sam68基因的子宫内膜癌细胞增殖情况
3 讨论
子宫内膜癌是常见的女性生殖系统恶性肿瘤之一。研究认为,子宫内膜细胞增殖机制失衡是引起内膜癌的重要因素,通过探索新型分子诊断标记和作用靶点,促进子宫内膜癌细胞的凋亡,被认为是当前子宫内膜癌研究的热点方向。子宫内膜癌的预后与细胞的分型分化密切相关,分化程度越高,细胞增殖和侵袭能力越弱,预后越好[6]。
近年来Sam68在肿瘤发生发展中的作用备受关注。早期的研究提示Sam68可能发挥类似抑癌基因的作用[7]。然而,近年更多的研究提示Sam68可能发挥癌基因的作用,与多种肿瘤的发生发展密切相关[8-10]。大量的实验观察表明,Sam68通过调节信号转导途径和基因表达调节直接参与肿瘤发生,特别是在内分泌相关癌症中[11]。Busa等[12]首先报道了Sam68在前列腺癌组织中相对于癌旁正常组织表达明显上调,下调Sam68可显著抑制前列腺癌细胞的增殖和抗凋亡能力;宫颈癌的研究[13]显示Sam68在宫颈癌组织中的表达明显高于正常宫颈组织,并能促进宫颈癌细胞增殖;Song等[14]报道了Sam68可能通过激活Akt/GSK-3β通路和抑制FOXO/p21/p27通路,促进细胞通过G1/S期检查点,从而促进乳腺癌细胞的增殖和恶性转化。 在卵巢中,Sam68表达在EOC组织和细胞系中上调,用小分子干扰RNA降低Sam68的水平可以将细胞周期阻滞在G1期从而损害细胞增殖[15]。
文献报道,Sam68基因敲除的小鼠表现出明显的子宫发育障碍[16],这使我们对Sam68在子宫内膜癌发生发展中可能起到的作用发生了兴趣。本研究选取了不同分化程度的子宫内膜癌细胞系 (HEC-1A, Ishikawa, RL952, JEC和AN3CA)及人正常子宫内膜组织中 Sam68 mRNA和蛋白表达水平进行检测,观察到Sam68在子宫内膜癌细胞系中的表达均明显上调,而在正常子宫内膜组织及癌旁组织中几乎检测不到Sam68基因及蛋白,这提示Sam68与子宫内膜癌的发生发展密切相关,进一步支持了本课题Sam68可能是子宫内膜癌癌基因的假设。本研究结果还发现,低分化AN3CA及中分化HEC-1A细胞中的Sam68 mRNA及蛋白水平明显高于高分化的Ishikawa细胞,提示Sam68的表达与子宫内膜癌细胞分化与恶性程度呈密切相关。这与Wang等[17]报道的子宫内膜癌患者中Sam68的高表达与组织学分级、FIGO分期和肌层侵犯有关相一致。
在上述基础上,我们对Sam68蛋白与肿瘤增殖相关的一些重要生物学特征进行了研究。本研究通过将Sam68的靶向siRNA转染至子宫内膜癌HEC-1A、RL952细胞中,经Western blot和qRT-PCR实验检测, 发现与对照组相比,Sam68蛋白和mRNA表达水平均明显受到抑制,该结果表明:Sam68RNAi能有效降低HEC-1A、RL952细胞中内源性Sam68基因的表达,为后续实验提供了研究基础。而将Sam68 cDNA转染至低表达Sam68的Ishikawa细胞后,Sam68蛋白和mRNA表达水平明显上升。在细胞增殖功能方面,在MTT法检测中发现,随着细胞培养时间的延长,Sam68-siRNA 和 Sam68- siRNA组细胞的增殖能力较各个对照组明显降低,过表达Sam68则促进增殖;平板克隆以及软琼脂克隆结果均证实,沉默Sam68 基因的表达可导致肿瘤细胞生存能力、集落形成能力降低,过表达Sam68则效果相反。
本研究结果表明,在子宫内膜癌细胞中抑制 Sam68 蛋白的表达,可抑制细胞增殖,但是具体机制尚未明确。后续研究中须对Sam68在子宫内膜癌发生发展中的机制作进一步探究,以期为子宫内膜癌的分子靶向治疗提供新的思路。
