AlN-MOCVD生长中气相寄生反应路径
2020-11-03仲婷婷
仲婷婷, 左 然
(江苏大学 能源与动力工程学院, 江苏 镇江 212013)
AlN是一种重要的第3代半导体材料,在3种主要的氮化物半导体(AlN,GaN,InN)中具有最宽的能带间隙(6.2 eV),因此特别适合制备紫外光电器件的阻挡层,在医学消毒、高效白光照明、数据存储等领域有巨大的应用前景[1-3].金属有机化学气相沉积(MOCVD)是制备AlN薄膜的主要方法.在AlN-MOCVD生长中,三甲基铝(Al(CH3)3,TMAl)与氨气(NH3)在载气(H2/N2)的携带下进入反应室,利用高温衬底引发化学反应,产生薄膜沉积.但在实际生长中,TMAl与NH3两种反应前体之间存在强烈的气相寄生反应,会形成纳米颗粒,导致生长效率降低以及薄膜质量下降[4-6].研究AlN的气相寄生反应路径对提高薄膜生长速率和质量至关重要.
前人研究[4-7]发现,在AlN-MOCVD生长中,TMAl和NH3在气相中一经混合,立即生成路易斯酸碱加合物TMAl:NH3.然后在较高温度,加合物脱去CH4生成氨基物DMAlNH2.氨基物活性很大,易聚合成多聚物[DMAlNH2]n(n≥2),最终形成纳米粒子.通过对AlGaN-MOCVD输运-反应过程的数值模拟以及对流动管反应器的试验测量,T. G. MIHOPOULOS等[4]首次提出AlN的MOCVD生长主要由加合路径控制,产生的二聚物为薄膜生长的主要前体,而三聚物为纳米粒子的主要前体.J. R. CREIGHTON等[5]利用原位激光散射测量,发现当载气由H2变为N2时,AlN的生长效率几乎不变,从试验上证明,加合路径是纳米颗粒形成的主要原因.A. V. LOBA-NOVA等[7]利用试验和数值模拟相结合的方法,得出AlN的生长速率随NH3流量的增加而降低,说明寄生反应随NH3流量的增加而增强.ZUO R.等[8]研究了TMAl气相加合反应的后续路径,发现含Al加合物或氨基物与第2个NH3继续发生加合反应在热力学上不利于发生.但是,含Al氨基物及其衍生物与NH3直接碰撞脱去CH4的双分子反应则有可能发生,而且通常具有较低能垒[9].
尽管对于MOCVD生长AlN的气相反应有了基本的了解,但是,对于TMAl加合反应的后续路径,特别是含Al氨基物形成多聚物后的路径走向目前仍不清楚,它们决定了纳米粒子的形成,进而影响薄膜生长的速度和质量.文中利用量子化学的密度泛函理论(DFT),对低聚物 [DMAlNH2]2,[MMAlNH]2,[MMAlNH]3与NH3的反应进行计算,对比不同温度下反应前后的吉布斯自由能差ΔG以及反应过渡态的活化自由能ΔG*,判断反应发生的路径和概率,进一步探索AlN-MOCVD气相寄生反应的机理.
1 计算模型和验证
根据前人的研究[4-9]结果,TMAl和NH3很容易通过加合反应和分子内反应生成氨基物DMAlNH2,其后的氨基物反应将存在2条路径:一条路径是在高温激活下氨基物与NH3发生双分子反应,一步步脱去CH4后得到Al(NH2)3;另一条路径是氨基物聚合成2种低聚物[DMAlNH2]2和[DMAlNH2]3,二者又可以通过分子内反应消去CH4,得到新的低聚物[MMAlNH]2和[MMAlNH]3.气相分子Al(NH2)3,[MMAlNH]2,[MMAlNH]3将不能经过分子内反应继续分解,因此很可能是提供表面反应或纳米粒子的直接前体.但是,由于MOCVD反应器中有大量过余的NH3,因此上述低聚物仍会与NH3发生双分子碰撞,文中将证明上述双分子反应的结果,进而确定AlN生长最可能的气相反应路径,以及最稳定的纳米粒子前体.由于前人[4-5]已经证实[DMAlNH2]3会导致纳米粒子的产生,因此文中仅考虑[DMAlNH2]2,[MMAlNH]2,[MMAlNH]3与NH3的反应.图1为MOCVD生长AlN可能的气相反应路径,特别包括二聚物和三聚物继续与NH3碰撞,脱去CH4后的结果.这些反应路径前人尚未研究过,是文中的研究重点.
采用Gaussian 09软件包[10]进行量子化学计算,选择密度泛函理论中的B3LYP/6-31G(d)方法,对含Al—N键的反应物、产物以及过渡态(TS)进行结构优化和频率计算.对过渡态结构进行内禀反应坐标计算(IRC)验证.上述计算方法已经在前人的研究[8-9]中得到证明.在不同的反应温度(298.15,573.15,873.15,1 273.15,1 473.15 K)下,采用B3LYP/6-311G(d,p)方法,对优化后的分子进行能量计算.通过对比不同温度下反应前后的自由能差ΔG,判断反应进行的方向.若ΔG<0,则反应自发进行;若ΔG>0,则反应难以发生.通过分析过渡态的活化自由能ΔG*,判断反应发生的速率.反应速率常数k∝e-ΔG*/(RT),因此,ΔG*越大,则反应速率越慢,或反应越难进行.
首先对AlN-MOCVD气相反应中的相关多聚物分子进行结构优化计算,并与文献值对比.优化后的多聚物分子构型如图2所示.

图1 AlN-MOCVD中气相反应路径示意图(框中为文中考虑的路径)
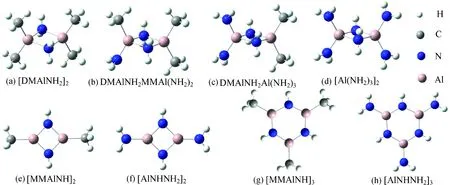
图2 AlN-MOCVD多聚物反应的主要分子优化结构示意图
图2中DMAlNH2具有C2V对称结构,其Al,C,N原子都在同一个平面内,C—Al—C的角度为123.56°,Al—N键长为0.178 9 nm,与文献[11]中得出的计算值123°,0.179 nm相符.其二聚物[DMAlNH2]2为环状结构,其中N—Al—N的角度为86.79°,Al—N键长为0.199 0 nm,与文献[12]中得出的计算值87.3°,0.197 5 nm基本相符.对于其他多聚物分子,由于前人研究很少,将通过DFT计算进行预测.
2 结果与讨论
2.1 [DMAlNH2]2与NH3的反应
如前所述,AlN-MOCVD气相中的氨基物DMAlNH2很容易聚合成二聚物.当二聚物[DMAlNH2]2生成后,可能与气相中大量存在的NH3依次发生双分子反应,脱去4个CH4,最终变成[Al(NH2)3]2,反应式为
[DMAlNH2]2+NH3=TS1=
DMAlNH2MMAl(NH2)2+CH4,
(1)
DMAlNH2MMAl(NH2)2+NH3=TS2=
DMAlNH2Al(NH2)3+CH4,
(2)
DMAlNH2Al(NH2)3+NH3=
TS3=MMAl(NH2)2Al(NH2)3+CH4,
(3)
MMAl(NH2)2Al(NH2)3+NH3=
TS4=[Al(NH2)3]2+CH4.
(4)
在上述反应中,环状二聚物[DMAlNH2]2与一个NH3分子碰撞,其中一个甲基CH3被氨气中的NH2取代,同时脱去CH4,变成DMAlNH2MMAl-(NH2)2,即反应(1);接着继续与另一个NH3分子碰撞,发生类似的取代反应,直到全部甲基被NH2取代,得到产物[Al(NH2)3]2,即反应(2)-(4).图3为在不同温度下[DMAlNH2]2与NH3连续反应的吉布斯自由能差变化.

图3 [DMAlNH2]2与NH3反应路径的吉布斯自由能差
由图3可见,反应(1)-(4)在计算温度范围内均有ΔG<0,即反应可自发进行.但由于反应(1)-(4)的活化自由能ΔG*随温度迅速增大,反应速率在高温时反而减小.
当考虑[DMAlNH2]2还可能发生消去CH4的分子内反应时,该反应显然跟[DMAlNH2]2与NH3的双分子反应存在竞争机制.[DMAlNH2]2的分子内反应为
[DMAlNH2]2=TS5=DMAlNH2MMAlNH+CH4,
(5)
DMAlNH2MMAlNH=TS6=[MMAlNH]2+CH4.
(6)
图4给出[DMAlNH2]2发生分子内反应的吉布斯自由能变化.

图4 [DMAlNH2]2分子内消去CH4反应路径的吉布斯自由能差
由图4可见,对于反应(5),当T<573.15 K时,ΔG>0,当T>873.15 K时,ΔG<0;利用线性插值法求得,当温度T>749.18 K时,ΔG<0,反应自发进行.对于反应(6),在所有的温度范围内,ΔG<0,反应自发进行.因此当温度T>749.18 K时,将有利于[DMAlNH2]2分子内反应的发生.
综合以上分析,当温度T<749.18 K时,只存在双分子反应路径,即方程(1)-(4),产物为[Al-(NH2)3]2.在温度T>749.18 K时,双分子反应(1)和分子内反应(5)均可自发进行,故二者存在竞争机制.反应(5)的活化自由能ΔG*=49.09~52.71 kcal·mol-1,且ΔG*随温度变化不大.而反应Al的活化自由能ΔG*=59.8~104.0 kcal·mol-1,远大于前者.因此在MOCVD反应器的高温环境下,[DMAlNH2]2分子内反应速率将明显快于双分子反应速率,因此反应趋向于活化自由能更低的分子内反应,经过两步消去CH4,生成[MMAlNH]2.
2.2 [MMAlNH]2与NH3的反应
根据前人研究[9]可知,[MMAlNH]2不能通过分子内反应继续分解(ΔG>0).但[MMAlNH]2与NH3碰撞后,可能发生脱去CH4的双分子反应,反应式为
[MMAlNH]2+NH3=
TS7=MMAlNHAlNHNH2+CH4,
(7)
MMAlNHAlNHNH2+NH3=
TS8=[AlNHNH2]2+CH4.
(8)
在上述反应中,[MMAlNH]2中的甲基CH3依次被氨气中的NH2取代,脱去两个CH4,最后得到新的环状二聚物[AlNHNH2]2.图5为在不同温度下[MMAlNH]2与NH3连续反应的吉布斯自由能差.

图5 [MMAlNH]2与NH3反应路径的吉布斯自由能差
由图5可见,反应(7)-(8)在计算温度范围内均有ΔG<0,反应可自发进行.反应(7)和(8)存在两个过渡态TS7,TS8,活化自由能ΔG*均为22~63 kcal·mol-1.随着温度的升高,活化自由能ΔG*明显增大,而由于温度引起的热能因子在1 000 K时可估算为RT=2 kcal·mol-1(R为气体常数),远小于活化自由能ΔG*的增加,因此反应速率主要取决于活化自由能ΔG*.由此得出结论,[MMAlNH]2和NH3可以经两步脱去CH4,最终生成[AlNHNH2]2,成为提供表面反应和纳米颗粒的前体.但随着温度的升高,活化自由能ΔG*增加,反应速率降低,因此反应倾向于在较低温度下进行.
2.3 [MMAlNH]3与NH3的反应
由于环状三聚物[MMAlNH]3也不能通过分子内反应继续分解(ΔG>0)[9],因此考虑该物质与NH3发生碰撞,脱去CH4的双分子反应的可能性,反应式为
[MMAlNH]3+NH3=
TS9=[MMAlNH]2AlNHNH2+CH4,
(9)
[MMAlNH]2AlNHNH2+NH3=
TS10=MMAlNH[AlNHNH2]2+CH4,
(10)
MMAlNH[AlNHNH2]2+NH3=
TS11=[AlNHNH2]3+CH4.
(11)
与[MMAlNH]2的双分子反应类似,[MMAlNH]3中的甲基CH3依次被氨气中的NH2取代,脱去3个CH4,最后得到新的环状三聚物[AlNHNH2]3.图6为在不同温度下[MMAlNH]3与NH3连续反应的吉布斯自由能差.

图6 [MMAlNH]3与NH3反应路径的吉布斯自由能差
由图6可见,反应(9)-(11)在计算温度范围内同样有ΔG<0,故反应可自发进行.反应(9)-(11)均存在过渡态,各反应的活化自由能ΔG*随温度的增大明显增大.因此在反应腔高温区,反应速率减慢,产物[AlNHNH2]3的浓度随温度升高而降低.由于没有其他平行反应路径,因此在AlN-MOCVD的气相中可能生成稳定的[AlNHNH2]3,该物质同样提供表面反应和纳米颗粒的前体.
3 结 论
利用量子化学的密度泛函理论(DFT),针对AlN-MOCVD气相中的含Al低聚物[DMAlNH2]2,[MMAlNH]2,[MMAlNH]3与NH3的寄生反应进行计算分析.研究发现,当温度T<749 K时,[DMAlNH2]2倾向于走与NH3的双分子反应路径,产物为[Al(NH2)3]2;当温度T>749 K时,[DMAlNH2]2同时存在分子内反应和双分子反应路径.分子内反应活化自由能更低,因此反应倾向于分子内消甲烷生成[MMAlNH]2.低聚物[MMAlNH]2,[MMAlNH]3与NH3发生双分子反应均有ΔG<0,即都可继续发生消甲烷反应,生成更为稳定的气相产物.因此得出结论,[Al(NH2)3]2,[AlNHNH2]2,[AlNHNH2]3可能是AlN-MOCVD气相反应中的末端粒子,也是提供表面反应和纳米颗粒的重要前体.
