30%咪鲜·氟环唑微乳剂高效液相色谱分析
2020-11-02张刚应
王 娟,张刚应
(河北中保绿农作物科技有限公司,北京 100193)
30%咪鲜·氟环唑微乳剂是一种具有预防和防治效果的广谱混配杀菌剂,咪鲜胺和氟环唑均执行企业标准分析方法。有关咪鲜胺分析方法已报道气相色谱法[6-9]、液相色谱法[3-5],氟环唑用液相色谱法分析也有报道[1-2]。但两者复配制剂的分析方法目前尚未见公开报道。本文所报道分析方法是采用高效液相色谱法对咪鲜胺和氟环唑混剂同时进行分离测定,提高了检测速度,拓宽了分析方法,简便且易行,适于生产质量控制及相关分析的研究。
1 实验部分
1.1 试剂和溶液 乙腈:色谱级;甲醇:色谱级;水:新蒸二次蒸馏水;氟硅唑标样:已知质量分数,w≥99.0%;咪鲜胺标样:已知质量分数,w≥99.0%;
1.2 仪器 高效液相色谱仪:water 2695带可调波长紫外检测器;色谱工作站;色谱柱:150mm×4.6mm(i.d.)不锈钢柱,内填SunFireTMC18、5μm填充物;过滤器:滤膜孔径约0.45μm;微量进样器:50μL。
1.3 色谱条件 流动相:甲醇+乙腈+水=30+40+30(V/V)使用前经0.45μm聚四氟乙烯膜过滤,并超声脱气20min;柱温:室温;检测波长:210 nm;流量:1.0mL/min;进样量:5μL。保留时间:咪鲜胺,10.6min;氟环唑,6.4min。典型色谱图(图1、2)。

图1 标样液相色谱图图2 试样液相色谱图
1.4 溶液的配制
1.4.1 标样溶液的制备 分别称取咪鲜胺标样0.1g、氟环唑标样0.05g(准确至0.000 2g)于100mL容量瓶中,用甲醇溶解,超声振荡5 min,冷却至室温,用甲醇稀释至刻度,摇匀。用移液管移取上述溶液5mL于50mL容量瓶中,用甲醇稀释至刻度,摇匀。
1.4.2 试样溶液的制备 称取30%咪鲜·氟环唑微乳剂试样0.5g(准确至0.000 2g)于100mL容量瓶中,用甲醇溶解并稀释至刻度,摇匀。用移液管移取上述溶液5mL于50mL容量瓶中,用甲醇稀释至刻度,摇匀。
1.4.3 测定 在上述操作条件下,待仪器基线稳定后,连续注入数针标样溶液,待相邻2针的峰面积相对变化<1.5%,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进行测定。
1.4.4 计算 将2针标样溶液和2针试样溶液中咪鲜胺(或氟环唑)的峰面积分别进行平均,试样中咪鲜胺(或氟环唑)的质量分数X(%)按式(1)计算:
(1)
式中:
X——试样溶液中咪鲜胺(或氟环唑)质量分数,%;
A2——试样溶液中咪鲜胺(或氟环唑)峰面积的平均值;
m1——咪鲜胺(或氟环唑)标样的质量,g;
P——咪鲜胺(或氟环唑)标样的质量分数,%;
A1——标样溶液中咪鲜胺(或氟环唑)峰面积的平均值;
m2——试样的质量,g。
计算结果应表示至2位小数。
2 结果与讨论
2.1 检测波长的选择 据相关研究显示,氟环唑在205nmnm[1-2]附近具有强吸收,咪鲜胺在210nm[3-5]附近有强吸收。但考虑到两者均需有较好的吸收,而且各种杂质不影响主组分的测定,因此,综合考虑多种因素且经实验验证后,选择210nm作为本方法的最佳吸收波长。
2.2 流动相的选择 根据咪鲜胺和氟环唑的特点,选用甲醇溶解样品。分别用甲醇-水、乙腈-水以及甲醇-乙腈-水按不同的体积比做了多个流动相配比试验。结果表明,当甲醇︰乙腈:水为30∶40∶30 (V/V)时,各组分均能得到较理想的分离,且基线平稳,峰形对称。
2.3 分析方法的线性相关性 分别配制5个不同浓度的标样溶液,按1.3色谱操作条件下进行分析,以咪鲜胺(氟环唑)浓度为横坐标,峰面积为纵坐标,绘制标准曲线,得到咪鲜胺和氟环唑线性方程结果(表1)。

表1 方法的线性回归方程实验
2.4 方法的精密度 称取同一批次30%咪鲜·氟环唑微乳剂6个样品,在1.3色谱操作条件下进行分析,分别获得咪鲜胺和氟环唑的标准偏差和变异系数,结果(表2)。咪鲜胺和氟环唑标准偏差分别为0.098、0.087,变异系数分别为0.49%、0.86%。
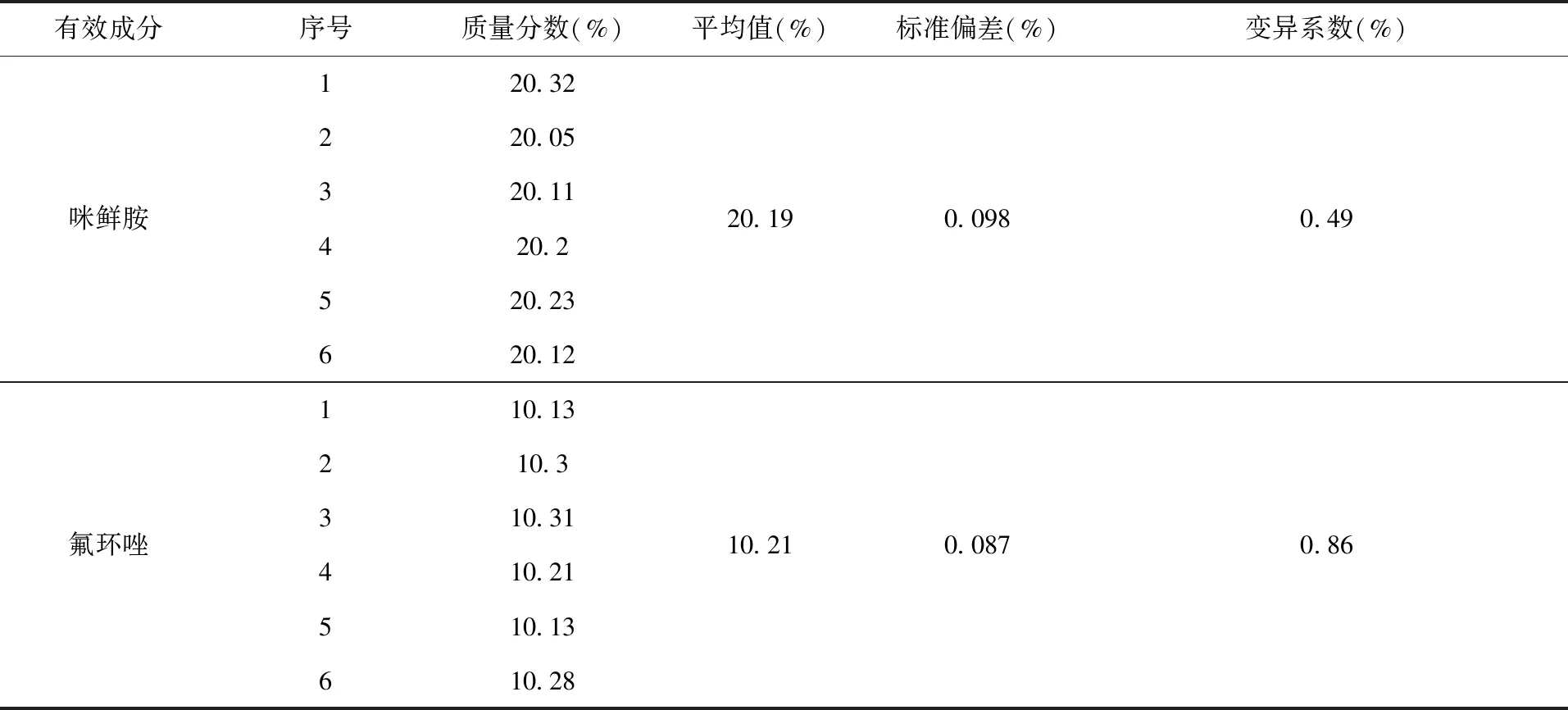
表2 方法的精密度实验
2.5 方法的准确度实验 按照配方比例要求,将除原药以外的所有助剂混匀,作制剂空白,按照制剂标称值在制剂空白中添加有效成分,合成5个制剂样品,在1.3色谱操作条件下进行分析,测定咪鲜胺的平均加标回收率为99.18%;氟环唑的平均加标回收率为99.53%(表3)。
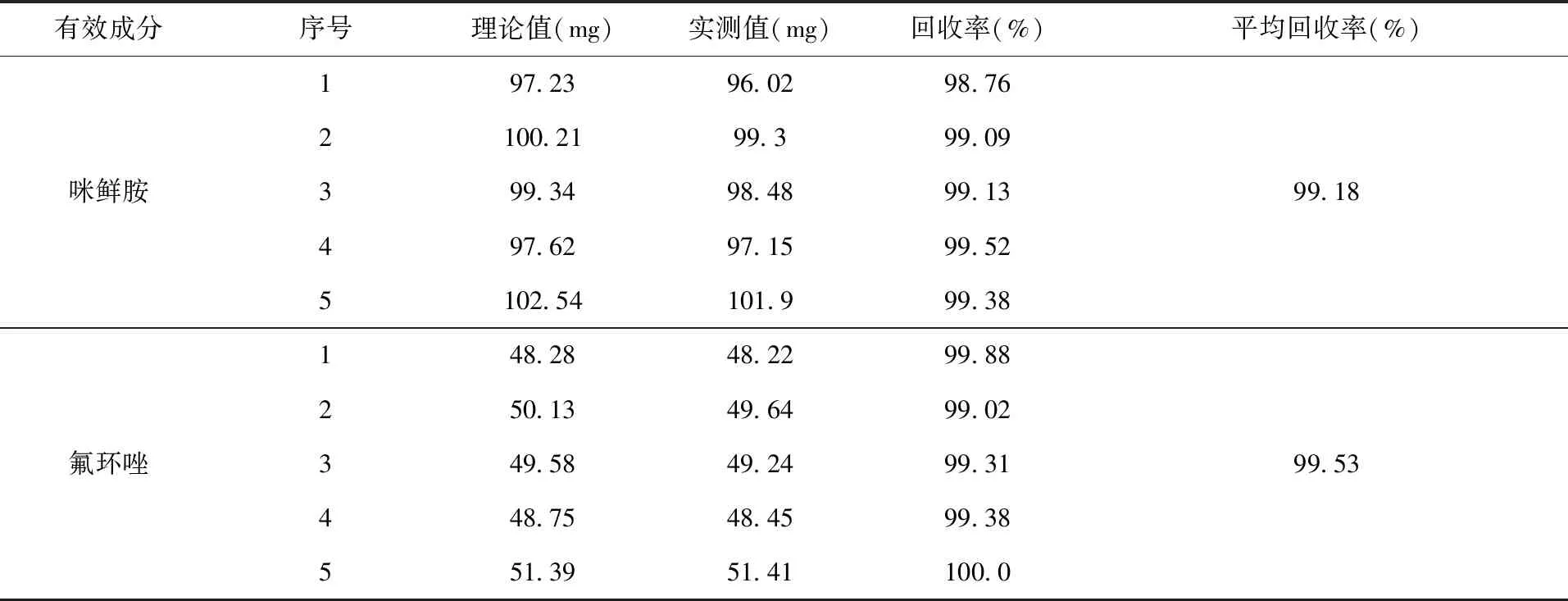
表3 方法的准确度实验
3 结论
本文所提出的采用高效液相色谱法对30%咪鲜·氟环唑微乳剂中咪鲜胺、氟环唑的含量进行分析,分离效果好,精密度和准确度高,可作为咪鲜胺和氟环唑混合制剂的有效分析方法。
