模拟蒸馏快速测定石脑油馏程及蒸气压
2020-10-28李玉萍李茂生时自立刘亚红宴红梅
李玉萍 , 李茂生 , 时自立 , 刘亚红 , 宴红梅
(中国石化 洛阳分公司 , 河南 洛阳 471012)
在炼油工业中,蒸馏过程广泛应用于分离工艺,中间产品的馏程是炼油工艺的重要控制和考核指标。目前石油产品馏程测定方法有恩氏蒸馏法和色谱模拟实沸点蒸馏法。恩氏蒸馏法为经典分析方法,该方法测定得到是各馏出体积时的混合物平均沸点,耗时长,油样消耗多,人工劳动量较大,最高可测定沸点365 ℃。色谱模拟蒸馏法是运用气相色谱技术模拟实沸点蒸馏法来测定石油和石油产品的馏程,测定时间短,自动化程度高,数据准确,能实现连续自动多次测定,最高可测定沸点达750 ℃,该方法多用于中间产品馏程测定。色谱模拟蒸馏数据除可用于加工过程控制、原油调配方案制定、常减压塔拔出率评价外,可间接测定石油产品除馏程以外的物性指标如润滑油的挥发度、相对分子质量、汽柴油机油中轻油稀释量等。对于工艺过程故障判断还具有恩氏蒸馏难以实现的效果,如轻组分中串入重组分,因馏分断层可造成恩氏蒸馏法终馏点判断提前,但可通过色谱模拟蒸馏技术观察色谱谱峰的分布变化加以判断[1-3]。
目前,国际标准ASTM D2887规定了初馏点高于55.5 ℃,终馏点低于538 ℃的石脑油、煤油、柴油、润滑油或其混合组分的测定方法;ASTM D7096规定了用色谱模拟蒸馏法测定沸点低于280 ℃样品馏程的方法;石化标准SH/T 0558-1993(2004)规定了用气相色谱仪测定石油馏分沸程分布,但仅适用于蒸气压低到能在室温下进样的馏分,不适用于石脑油或石脑油组分的测定。本文拟通过调整优化试验条件,使馏程为30~200 ℃的石脑油能通过色谱模拟蒸馏法测定其馏程。
除馏程外,蒸气压是石脑油的另一重要物理性质,蒸气压越高说明液体越容易气化。蒸气压除用于判断其蒸发性的大小以外,还用于判断其在使用时形成气阻的倾向。蒸气压越大,形成气阻的倾向就越大;为充分利用色谱数据信息,减少分析样品量、分析时间,近年来,国内外的研究工作者均在探讨馏程数据与油品的一些物理性质相关联,从而获得油品的其它物性数据,且已取得了一些研究成果[4]。本文通过应用多元线性回归方法,对石脑油的模拟馏程数据与其蒸气压建立关联,用于中间控制分析,减少了过程分析的工作量[5-6]。
1 实验部分
1.1 实验原理
色谱模拟蒸馏技术是基于样品中单个组分在色谱柱上按照沸点顺序流出的原理,采用具有一定分离效果的非极性色谱柱,在线性程序升温条件下,测定已知正构烷烃的保留时间,通常选择沸点覆盖所测样品沸程的正构烷烃混合物。在与样品相同的色谱测定条件下,在色谱柱内产生分离,在谱图上形成分离的色谱峰,通过判断其保留时间并结合该组分的沸点,得到沸点与保留时间的关系,建立沸点与保留时间关联图。在该色谱条件下,样品各组分按沸点次序分离,同时对色谱图进行切片积分,首先计算出样品谱图总面积和不同保留时间内的谱图面积,再计算出面积百分数与保留时间的关系,即百分收率与保留时间的关系,然后与沸点-保留时间的关系进行关联,得到百分收率与温度的关系,即馏程数据。在FID检测器上石油烃类化合物响应值与质量成正比,故相对质量校正因子都比较接近,认为是试样的质量百分含量。定量方法有归一化法、内标法、增量内标法等,对于全部出峰的轻质油品采用归一化法进行定量。研究人员通过大量的试验研究,采用多元线性回归和最小二乘法多项式回归分析,逐步建立起色谱模拟蒸馏数据与恩氏蒸馏数据之间的关联模型,通过关联计算转化成不同回收体积下对应的温度即可获得与GB/T 6536 具有可比性的恩氏馏程结果[7]。
恩氏蒸馏法按照GB/T 6536标准,将100 mL油品放到标准的蒸馏烧瓶中,按照规定蒸馏速度蒸馏,馏出第一滴冷凝液时的气相温度称为初馏点。温度逐渐升高,依次记录馏出10 mL、50 mL直至95 mL时的气相温度,得到对应体积百分率的温度。当气相温度升高到一定数值后就不再上升,其最高气相温度被称为终馏点。
色谱模拟蒸馏与恩氏蒸馏的初馏点和终馏点定义存在不同之处,模拟蒸馏的初馏点为达到色谱峰总净面积0.5%时对应的温度,终馏点为达到色谱峰总面积99.5%对应的温度,其它收率点的温度为从1%~99%(质量百分比)对应面积整数百分数的时间,通过正构烷烃沸点-保留时间关联图,采用线性内插方法求出对应的温度。
1.2 仪器与试剂
1.2.1仪器
Agilent6890气相色谱仪;FID检测器;中石化科学研究院D2887模拟蒸馏软件;色谱柱:10 m×0.53 mm ×0.5 μm非极性色谱柱 、Aglient7683自动进样器、PTV进样口。
1.2.2 试剂
NC5~NC16标样、NC5~NC44标样,1#参考油。
1.3 实验条件选择
在色谱柱、载气、固定液液膜厚度等硬件配置一定的情况下,影响色谱分离度效果的主要影响因素有进样量、柱温、载气线速[8]。因采用程序升温模式,在固定初始温度的前提下,升温速率成为主要影响因素,因此以进样量、升温速率、载气线速三个主要因素进行条件筛选试验,其它次要因素则通过简单摸索提前设定。
因石脑油初馏点约35 ℃,进样时样品易挥发损失,故应将汽化室温度和色谱柱初温尽量低控。色谱柱初始温度设定为30 ℃,这样既可以减少因挥发引起的样品失真,又可能提高色谱分离效率,减少色谱柱固定液流失。进样量、升温速率、载气流速采用正交试验法进行筛选。气相色谱法测定石脑油馏程部分操作条件如下:进样方式,不分流;检测器温度300 ℃;汽化室温度,初温150 ℃,保持1 min,升温速率50 ℃/min,终温20 ℃,柱温初始温度30 ℃;载气,氮气;辅助气的流量(高纯氮)20 mL/min;燃气流量(高纯氢)为30 mL/min;助燃气流量(净化空气) 300 mL/min。
1.3.1试验因子水平设定
通过初步摸索,确定分流比、升温速率、载气流速的因子水平如表1所示。

表1 试验因子水平表
1.3.2正交试验结果
以溶解有1%(质量百分比)的正十六烷与正十八烷的正辛烷样作为试验样,以正十六烷与正十八烷的分离度作为试验的目标值,试验因子配比安排和试验结果如表2所示。

表2 正交试验安排及试验结果
从以上试验统计结果看出,三因子A、B、C分别取A3B1C1分离度最大,因此选择进样量为0.1 μL,升温速率40 ℃/min,载气流速6 mL/min。同时从各因子极差值得出进样量对分离度影响最大,载气流速影响最小。
1.4 空白分析
由于程序升温时基线有一定程度的漂移,因此需进行基线扣除,以获得准确的真实峰面积。在与选定的试验条件下,不进样品,启动升温程序,记录色谱基线信号,当信号积分峰面积小于样品峰面积0.3%时才能进行校正样或样品分析。正常情况下,空白分析基线应该平滑、无毛刺和残余峰、高温段时基线平稳无上翘或下行,否则应重新进行基线补偿分析,直到满足以上要求为止才能进行校正样分析或样品分析。在每次开机后均应进行空白分析,必要时在两次样品分析之间进行。
1.5 校正样分析
石脑油样馏程范围为35~205 ℃,由于石脑油中可能含有一定量的C4轻组分,但市售的标样中只有C5~C16,因此需要在C5~C16校正样中加入C4组分,以将石脑油组分全部覆盖在校正样的沸程之内。加入方法为:将C5~C16标样装入2 mL色谱小瓶中,用注射器吸入1 mL碳四组分注入该色谱小瓶中,该标样成为C4~C16校正样。在上述石脑油试验条件下进C4~C16校正样,得校正样沸点与保留时间对照表见表3,谱图见图1,并将保留时间与沸点作关联图见图2。

表3 C4~C16校正样沸点与保留时间对应表
1.6 样品分析
用石脑油在与其校正样相同的条件下进样,谱图见图1。

图1 石脑油模拟蒸馏色谱图
1.7 重复性考察
重复测定同一石脑油样7次,考察其重现性,如表4所示。

表4 重复性考察表
从表4可看出,用模拟蒸馏法测定的7次石脑油馏程结果的极差均在GB/T 6536 的重复性范围内,说明其测定的精密度能满足标准要求。
将模拟蒸馏结果与GB/T 6536实测值进行对照,考察其准确性,对比试验结果如表5所示。
从以上石脑油模拟蒸馏结果与GB/T 6536-2010测定值比较可看出,两方法测定结果一致,满足标准重复性要求,模拟蒸馏可代替GB/T 6536-2010进行石脑油的馏程测定。

表5 模拟蒸馏数据与恩氏蒸馏数据对照表 ℃
2 蒸气压预测
石脑油的蒸气压主要由C7以前组分的分压构成,利用模拟蒸馏所得的轻组分数据,将初馏点、5%馏出温度、10%馏出温度经多元线性回归处理与蒸气压建立关联[12]。将蒸气压设为因变量Y,初馏点、5%馏出温度、10%馏出温度分别设为X1、X2、X3自变量,建立方程Y=K+k1X1+K2X2+K3X3。
选定7个石脑油样品,用模拟蒸馏法测定其馏程,用GB/T 8017-2012(石油产品蒸气压测定法(雷德法))测定其蒸气压,将初馏点、5%馏出温度、10%馏出温度和蒸气压结果运用多元线性回归法计算出各个系数值。馏程与蒸气压数据见表6。
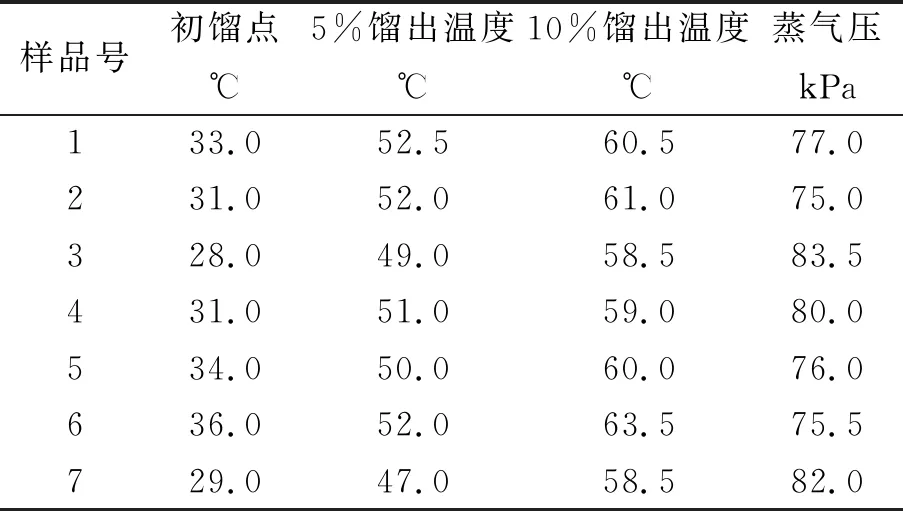
表6 馏程与蒸汽压数据对照表
通过计算得出:K=150.7、K1=-0.767、K2=-0.654、K3=-0.294。
因此关联公式为:Y=150.7-0.767HK-0.654T5%-0.294T10%
采用F检验法来检验该方程是否有效:n=7,fT=6,fR=3,fE=3,F1-α(fR,fE)=9.28,F=10.55,因此F>F1-α(fR,fE),说明该回归方程是显著的,线性关系良好。
用实测值与预测值进行对照,验证回归方程是否有效,见表7。
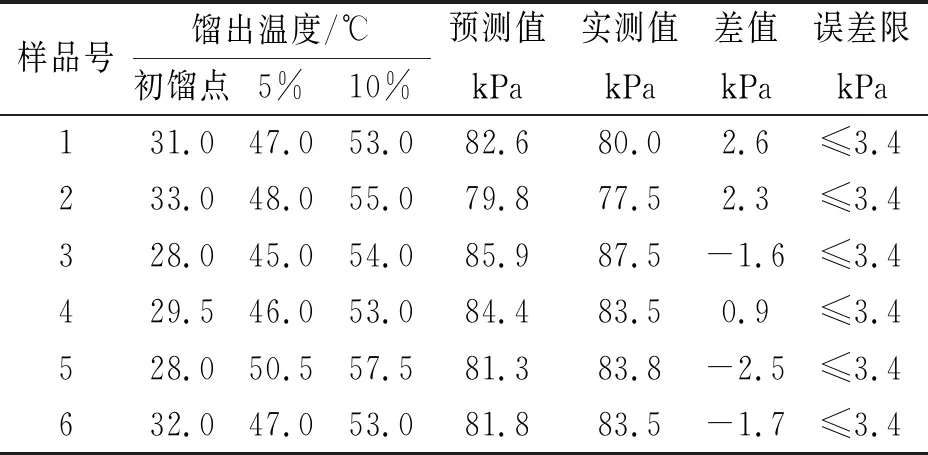
表7 实测值与预测值对照表
从以上预测值和实测值的对照可看出,用多元线性回归方程预测得出的蒸气压结果与实测值结果符合方法GB/T 8017-2012的重复性要求,说明用线性回归预测得到的蒸气压结果有效。
3 方法应用与讨论
3.1 色谱系统验证
3.1.1固定液流失
色谱柱使用一段时间后,因反复升温、降温,色谱柱固定相流失造成分离度下降、保留时间提前,但该色谱软件馏程计算是按照保留时间与峰面积切片积分的关联为依据,保留时间的漂移导致分析结果偏差,因此需要定期(每周至少1次)用参考油验证。当参考油馏程结果与其实际值超出再现性要求时,应该用校正样对其保留时间与沸点的模型进行校正。
当各为1%(质量百分比)的正十六烷与正十八烷分离度<3时,判断该色谱柱已失效柱,若色谱柱失效则将其掉头使用或切除一小段继续使用,若仍达不到要求则更换色谱柱。分离度具体检测方法:将各为1%(质量百分比)的正十六烷与正十八烷溶解在正辛烷中,在样品测定条件下进样,依据保留时间与半峰宽计算该两种物质的分离度。
由于固定液在高温下流失,可能造成FID离子头喷嘴周围形成晶状沉积物,从而影响其响应特性或堵塞喷嘴,影响点火,因此应定期清除沉积物。
进样衬管污染样品中的重组分容易污染色谱柱及离子头,某些重组分在进样瞬间附着在毛细管柱衬管上,造成进样不完全,影响分析结果准确性。因此应定期检查进样衬管并适时清洗、更换。
3.1.2柱接头漏气
由于经常升降柱温,色谱柱接汽化室及检测器的接头易松动,造成载气渗漏,使得部分轻组分遗失。初馏点后移,改变了馏程,影响分析结果的准确性,应关注柱头压的变化,柱头压减少时,及时检查柱接头气密性,保证系统气密性良好状态。
3.2 基线补偿
因为样品谱图积分采用切片积分的方式,因此必须扣除基线空白影响。通常影响色谱基线稳定的因素有柱流失、隔垫流失、载气流量不稳、系统气密性、程序升温基线漂移、重组分残留、噪声等,为消除对样品分析的影响,在每天进样前应进行空白分析,在与样品分析相同的色谱操作条件下重复操作,基线尾部高温段与前边低温段的基线平行且重复性好,则可作为正常空白样,否则认为空白样失真。
3.3 轻组分的影响
因校正样和石脑油样品中含有一定量的C3~C5轻组分,在室温下极易挥发损失造成样品失真,在进样量本来就很少的情况下,对分析结果特别是初馏点的影响非常明显,引起初馏点升高。在采样、保存、转移和进样过程中应防止组分挥发。在储存时应放置在冰中冷却,等到温度达到低温平衡之后再进样。进样小瓶在进样前同样放入冰箱中冷却,样品转移时动作应迅速,用自动进样器进样前不宜提前放置于进样盘中太久,防止样品放置过程中的挥发损失。应使色谱柱初始温度尽可能低于样品的初馏点,否则影响初馏点测定的精密度和准确度。
3.4 样品馏程范围的影响
因本方法采用毛细管分析柱,柱容量一定,且定量方法采用沸点与保留时间的对应关系而得,如果样品馏分较为集中,馏程范围较窄,高含量组分易造成色谱柱过载、保留时间发生位移、峰形畸变,导致计算结果产生偏差,当工艺条件发生变化或产品结构调整时,石脑油的馏程可能出现分段较集中的情况,对于此种情况应减小进样量。
3.5 进样速度的影响
由于模拟蒸馏方法计算的基础是保留时间与沸点的关联关系,保留时间的重复性是分析结果准确度的关键因素,因此对于进样速度的重复性要求很高,用于该方法的仪器应配置自动进样器。
3.6 模型数据的可靠性
因蒸气压与馏程的线性方程的建立基础是实测的蒸气压与模拟馏程结果,为了获得预测蒸气压结果与馏程结果的良好线性关系,在测定蒸气压时应注意轻组分的挥发,严格控制样品冷冻温度和水浴温度,样品不宜放置过久,应尽可能与模拟蒸馏测定同时进行。
4 结论
针对石脑油蒸气压偏高,挥发性大的问题,在未采用冷柱头进样方式的情况下,通过降低汽化室温度、色谱柱初始温度、冷却试样等措施,可以获得与GB/T 6536 一致的结果。试验条件的优劣决定色谱分离效果的好坏,本文采用正交试验法对色谱分离的主要参数进行筛选,以最少的试验次数获得了最佳的试验条件。利用油品性质之间的相关性,依据石脑油的蒸气压源自轻组分的含量高低,建立多元线性回归方程,在获得模拟蒸馏结果的情况下,采用“软测量”方式,通过线性回归方程预测石脑油的蒸气压,得到与GB/T 8017一致的结果。通过采用色谱模拟蒸馏技术和数理统计方法测定石脑油蒸气压、自动化程度高、耗时少、用样量少、准确度高、环境友好、提高了过程监控效率,可应用于炼油企业过程产品质量控制。
