HPLC同时测定感冒解毒灵颗粒中8种有效成分的含量
2020-10-22张秋芳刘宏明
张 娟,杜 武,张秋芳,刘宏明*
(1.淄博市食品药品检验研究院,山东 淄博 255086;2.淄博市中心医院,山东 淄博 255036)
感冒解毒灵颗粒收载于《卫生部药品标准中药成方制剂》第十九册,是纯中药制剂,由紫苏叶、板蓝根、前胡、防风、金银忍冬叶、连翘、麦冬、苦杏仁、羌活、川芎、陈皮、桑白皮、牛蒡子等13味中药构成,功能主治为解表散寒,宣肺止咳,清热解毒[1]。组方中,板蓝根有抗病毒、降血脂和降血糖等药理作用,(R,S)-告依春为代表的生物碱已被证明有明显的抗病毒活性[2];防风主治外感风寒、周身疼痛等症,升麻素苷和5-O-甲基维斯阿米醇苷等色原酮是防风主要的活性成分[3]。现原感冒解毒灵颗粒质量标准无含量测定项目,为了更有效地控制感冒解毒灵颗粒质量,结合中国药典,本文选取了感冒解毒灵颗粒中的6味药材的8种指标性成分作为研究对象,包括板蓝根中的(R,S)-告依春,防风中的升麻素苷和5-O-甲基维斯阿米醇苷,金银忍冬叶中的新绿原酸、绿原酸[4],连翘中的连翘苷,陈皮中的橙皮苷,牛蒡子中的牛蒡苷,建立了高效液相色谱法(HPLC)同时测定感冒解毒灵颗粒中8种成分含量的方法,为进一步完善其质量标准及制剂开发提供参考。
1 仪器与试药
1.1 仪器
Agilent 1260高效液相色谱仪,DAD检测器(美国安捷伦公司);KS-300E超声波清洗器(宁波科生仪器厂);十万分之一MS205DU电子天平(瑞士梅特勒-托利多)。
1.2 试药
(R,S)-告依春对照品(批号:111753-210706,含量100 %),绿原酸对照品(批号:110753-201817,含量96.8 %),升麻素苷对照品(批号:111522-201712,含量96.2 %),5-O-甲基维斯阿米醇苷对照品(批号:111523-201610,含量96.1 %),橙皮苷对照品(批号:110721-201617,含量96.1 %),连翘苷对照品(批号:110821-201615,含量94.9 %),牛蒡苷对照品(批号110819-201611,含量95.9 %)均购自中国食品药品检定研究院;新绿原酸对照品(ANPEL,批号:E0420010,含量99.0 %);感冒解毒灵颗粒(批号:181214,180902,181114)均为市购;乙腈为色谱纯,水为超纯水,其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件
色谱柱:SynergiTMHydro-RP 80Ȧ(250 mm×4.6 mm,4 μm);流动相:乙腈(A)-0.1 %磷酸的水溶液(B),梯度洗脱(0~15 min,5 % A;15~30 min,5 % A ~8 % A ;30~58 min,8 % A~12 % A;58~65 min,12 % A ~18 % A;65~75 min,18 %A~25 % A;75~90 min,25 % A~30 %A;90~100 min,30 % A~50 % A);流速:1.0 ml/min;柱温:35 ℃;检测波长:(R,S)-告依春245 nm,新绿原酸和绿原酸327 nm,升麻素苷和5-O-甲氧基维斯阿米醇苷254 nm,橙皮苷283 nm,连翘苷和牛蒡苷278 nm;进样量:10 μl。在此色谱条件下,8种成分理论板数均大于5000,相邻两色谱峰分离良好,见图1。
2.2 溶液的制备
2.2.1 供试品溶液 取感冒解毒灵颗粒的内容物,研细,取细粉约2 g,精密称定,置入具塞锥形瓶,精密加入甲醇20 ml,密塞,称定重量,超声提取(功率为300 W,频率为40 kHz)30 min,放冷,再称定重量,用甲醇补足减失的重量,摇匀,经0.45 μm的微孔滤膜过滤,取续滤液作为供试品溶液。
2.2.2 阴性样品溶液 根据感冒解毒灵颗粒处方,模拟其制备工艺,制备不含板蓝根、防风、金银忍冬叶、连翘、陈皮、牛蒡子的阴性样品,依照2.2.1项下方法制备相应阴性样品溶液。
2.2.3 对照品溶液 精密称取(R,S)-告依春对照品10.97 mg,置入100 ml量瓶,加甲醇适量,振摇使溶解,加甲醇稀释至刻度,摇匀,作为(R,S)-告依春对照品贮备液(0.1097 mg/ml);同法制成新绿原酸对照品贮备液(0.4035 mg/ml)、绿原酸对照品贮备液(0.9835 mg/ml)、升麻素苷对照品贮备液(1.045 mg/ml)、5-O-甲基维斯阿米醇苷对照品贮备液(0.6400 mg/ml)、连翘苷对照品贮备液(0.9604 mg/ml)、牛蒡苷对照品贮备液(1.100 mg/ml)。精密称取橙皮苷对照品16.99 mg,置入50 ml量瓶,分别精密加入上述7种对照品贮备液0.5,2,2,2,1,5,3 ml,加甲醇稀释至刻度,摇匀,作为混合对照品贮备溶液[含(R,S)-告依春1.097 μg/ml、新绿原酸16.14 μg/ml、绿原酸39.34 μg/ml、升麻素苷41.79 μg/ml、5-O-甲基维斯阿米醇苷12.80 μg/ml、橙皮苷326.5 μg/ml、连翘苷96.04 μg/ml、牛蒡苷绿原酸66.00 μg/ml]。精密量取10 ml混合对照品贮备液,置入20 ml量瓶,加甲醇稀释至刻度,摇匀,作为混合对照品溶液[含(R,S)-告依春0.5485 μg/ml、新绿原酸8.070 μg/ml、绿原酸19.67 μg/ml、升麻素苷20.89 μg/ml、5-O-甲基维斯阿米醇苷6.4 μg/ml、橙皮苷163.3 μg/ml、连翘苷48.02 μg/ml、牛蒡苷33 μg/ml]。
2.3 专属性试验
分别精密吸取对照品溶液、供试品溶液及阴性样品溶液各10 μl,注入液相色谱仪,记录色谱图,见图1。供试品溶液的色谱图中,在与对照品溶液的色谱图相应的位置上,有相同保留时间的色谱峰,而阴性样品溶液在此保留时间处无干扰。
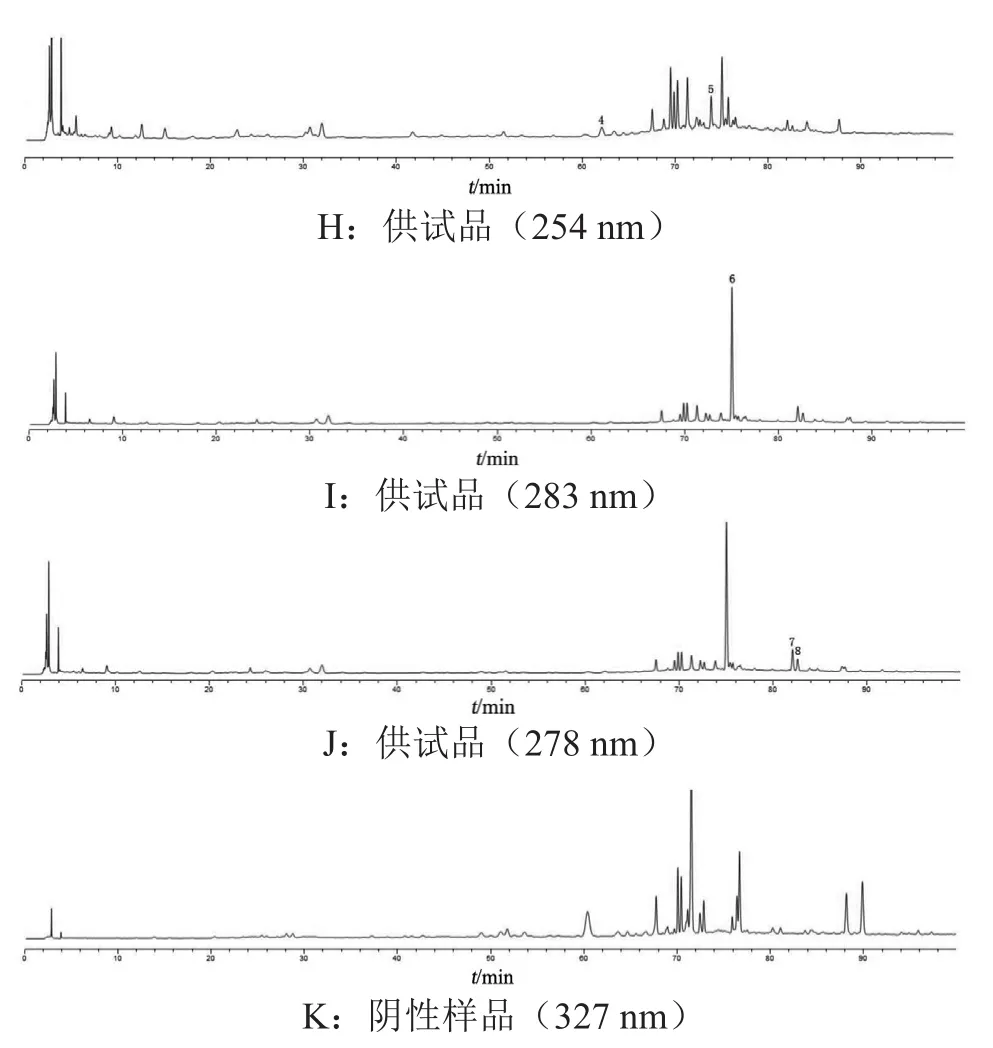
图1 HPLC色谱图
2.4 线性关系考察
分别精密吸取混合对照品溶液1,5,10,15,20 μl及混合对照品贮备液20 μl,按2.1项下色谱条件依次进样,记录8个成分的色谱图及峰面积。以进样量(μg)为横坐标,以峰面积为纵坐标,绘制标准曲线,计算回归方程,结果见表1。结果表明8个成分在表1范围内线性关系良好。
2.5 定量限和检测限
精密量取混合对照品溶液1 ml,置入50 ml量瓶,用甲醇稀释至刻度,摇匀;精密量取适量,用甲醇稀释制成不同浓度系列的混合对照品溶液,按2.1项下的色谱条件进行测定,以信噪比(S/N)为10时测定定量限,信噪比(S/N)为3时测定检测限,结果见表1。
2.6 精密度考察
取混合对照品溶液,按2.1项下色谱条件,连续进样5次,记录色谱图。(R,S)-告依春、新绿原酸、绿原酸、升麻素苷、5-O-甲基维斯阿米醇苷、橙皮苷、连翘苷和牛蒡苷峰面积RSD分别为1.20 %,0.73 %,0.92 %,0.85 %,1.32 %,1.15 %,0.71 %和0.92 %。结果表明,仪器精密度良好。

表1 8种成分线性关系考察结果及定量限、检测限
2.7 重复性考察
取同批号样品(批号:181214),按2.2.1项下方法,制备6份供试品溶液,分别测定(R,S)-告依春、新绿原酸、绿原酸、升麻素苷、5-O-甲基维斯阿米醇苷、橙皮苷、连翘苷和牛蒡苷含量,结果样品中平均含量分别为0.06243,0.1832,0.3634,0.4292,0.4507,12.21,3.214和1.836 mg/袋,RSD分别为1.37 %,1.03 %,1.14 %,1.16 %,0.81 %,0.62 %,1.45 %和1.18 %。结果表明该方法重复性良好。
2.8 稳定性考察
取按2.2.1项下方法制备的供试品(批号:181214)溶液,分别于制备后0,4,8,12,18和24 h进样分析,测得(R,S)-告依春、新绿原酸、绿原酸、升麻素苷、5-O-甲基维斯阿米醇苷、橙皮苷、连翘苷和牛蒡苷峰面积的RSD分别为1.14 %,0.84 %,0.98 %,1.12 %,1.35 %,0.91 %,1.34 %和1.23 %,结果表明,供试品溶液24 h内稳定性良好。
2.9 加样回收率实验
取已知含量的感冒解毒灵颗粒样品(批号:181214)6份,每份约1 g,精密称定,置入具塞锥形瓶中,分别精密加入混合对照品液[(R,S)-告依春、新绿原酸、绿原酸、升麻素苷、5-O-甲基维斯阿米醇苷、橙皮苷、连翘苷和牛蒡苷8种成分的浓度分别为1.207,3.632,7.868,8.358,8.960,241.0,63.32,385.0 μg/ml]10 ml,再精密加入甲醇10 ml,按2.2.1项下方法制备供试品溶液,分别测定8种成分含量,计算回收率。结果(R,S)-告依春、新绿原酸、绿原酸、升麻素苷、5-O-甲基维斯阿米醇苷、橙皮苷、连翘苷和牛蒡苷8种成分的平均回收率分别为99.76 %,99.43 %,99.90 %,97.84 %,99.62 %,99.25 %,98.67 %,98.10 %,RSD分别为1.80 %,1.04 %,1.59 %,1.77 %,1.73 %,0.58 %,1.49 %,1.50 %,结果见表2。
2.10 样品测定
取不同生产厂家感冒解毒灵颗粒,按2.2.1项下方法制备供试品溶液,照上述色谱条件测定,样品中(R,S)-告依春、新绿原酸、绿原酸、升麻素苷、5-O-甲基维斯阿米醇苷、橙皮苷、连翘苷和牛蒡苷的含量测定结果见表3。
3 讨论
3.1 提取方法的选择
考察了加热回流提取法和超声处理两种提取方法,提取率无显著差别,因此选择操作简便的超声提取法。同时,考察了3个不同提取时间(20,30和40 min)对提取效率的影响,30 min各成分的响应高于20 min,而40 min与30 min对各成分响应影响不大,最终确定了提取时间为30 min[5]。
3.2 波长的选择
感冒解毒灵颗粒成分复杂,单一检测波长会出现色谱峰组成单一、峰面积偏小、特征峰数量少等情况,本文采用DAD 检测器进行全波长扫描(200~400 nm),分析8种成分的紫外光谱,选择了8种成分的最佳检测波长[6-8]。多波长同时检测提高了检测成分的灵敏度,减少了测定误差。下一步开展感冒解毒灵颗粒的指纹图谱研究时可选用共有峰较多的245 nm作为指纹图谱的吸收波长。
3.3 流动相的选择
甲醇-水或乙腈-水[6]条件下的色谱峰拖尾,分离度和峰形较差,以乙腈-0.1 %磷酸为流动相时,峰形尖锐。采用等度洗脱[7-8]很难将8种成分与其他色谱峰完全分开,因此本文建立了一种梯度洗脱方法,色谱峰分离效果好,且8种成分均具有较好的稳定性、精密度和重复性,减少了测定误差。
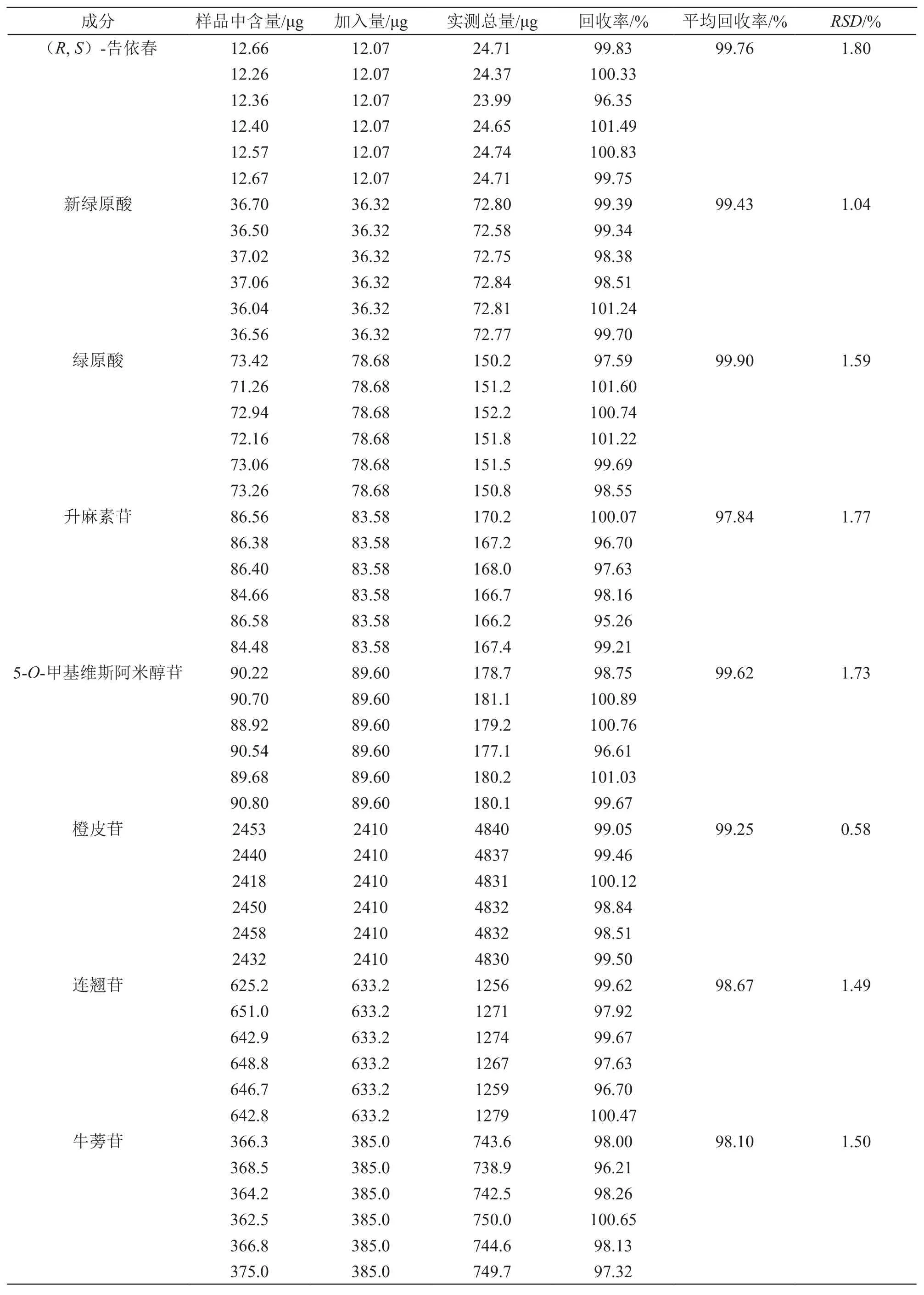
表2 回收率试验结果(n=6)

表3 样品含量结果/mg·袋-1
综上,本文建立HPLC法同时测定感冒解毒灵颗粒中8种成分的定量分析方法,方法重现性好,专属性强,可为综合评价感冒解毒灵颗粒提供参考。
