超高效液相色谱串联质谱法同时测定面粉中曲酸和噻二唑
2020-10-20林芳
林芳


摘 要:建立超高效液相色谱-串联四级杆质谱法(UPLC-MS/MS)同时测定面粉中曲酸和噻二唑的方法。色谱柱为ACQUITY UPLC? BEH C18(2.1 mm×100 mm,1.7 ?m),流动相为0.1%甲酸5 mmol·L-1甲酸铵水溶液∶乙腈,梯度洗脱,流速为0.45 mL·min-1。采用多反应监测模式(multiple reaction monitoring,MRM),曲酸为ESI正电离源,定量离子对140.3/69;噻二唑为ESI负电离源,定量离子对130.8/57.9。样品经纯水超声提取30 min,过膜后直接测定,外标法定量。结果表明,曲酸在1.00~50.00 ?g·L-1,噻二唑在10.00~500.00 ?g·L-1范围内线性关系良好,相关系数≥0.995。面粉样品中3个浓度加标水平即曲酸含量0.10、0.20和0.40 mg·kg-1,回收率为100.00%~105.00%,相对标准偏差为2.50%~10.72%;噻二唑含量1.00、2.00和4.00 mg·kg-1,回收率为86.00%~89.00%,相对标准偏差为3.65%~9.97%。该方法操作简便、回收率高,可用于大批量面粉中曲酸和噻二唑的同时测定。
关键词:面粉;曲酸;噻二唑;超高效液相色谱串联质谱法
Abstract:A new method was developed for the determination of Kojic acid (KA) and 2-mercapto-5-methyl-1,3,4-thiadiazde (MMTD) in flour by ultra high performance liquid chromatography-tandem mass spectrometry/mass spectrometry. The chromatography column was ACQUITY UPLC? BEH C18 (2.1 mm×100 mm, 1.7 ?m), mobile phases were 0.1%formic with 5 mmol·L-1 ammonium formate in water solution and acetonitrile, with gradient elution, the flow rate was 0.45 mL·min-1. Using multiple reaction monitoring (MRM) mode, the ESI mass spectrometry was positive ion source for KA, 143.1/69 and negative ion source for MMTD, 130.8/57.9. The sample was extracted by water solution, the quantitation was carried out with the external standard method. The linear range of the method was 1.00~50.00 ?g·L-1 for KA and 10.00~500.00 ?g·L-1
for MMTD, with a correlation coefficient not less than 0.995. The recoveries in flour matrix at the three spiked levels of 0.10, 0.20, 0.40 mg·kg-1 for KA and 1.00, 2.00,4.00 mg·kg-1 for MMTD, were in the range of 100.00%~105.00% for KA and 86.00%~89.00% for MMTD, and the relative standard deviations were in the range of 2.50%~10.72% for KA and 3.65%~9.97% for MMTD. The method is simple, high recoveries and is suitable for the simultaneous confirmation and quantificaition of KA and MMTD residue in flour which applies in batch detection.
Key words:Flour; Kojic acid; 2-mercapto-5-methyl-1,3,4-thiadiazde (MMTD); UPLC-MS/MS
中圖分类号:TS207.3
曲酸(Kojic acid,KA),化学名为5-羟基-2-羟甲基-1,4-吡喃酮,易溶于水、乙醇和丙酮,1907年由斋藤从蒸米发酵物中发现[1]。曲酸具有多种生物活性,如抑制酪氨酸酶活性[2-3],广泛在化妆品和药物制剂中使用[4],减少黑素的形成。聂西度等[5]表示曲酸分子中的羟基可以使人体内积累游离基有致癌性。陈永红等[6]以小鼠为研究对象,发现体外试验在一定时间内高质量浓度曲酸可以导致DNA损伤。因此曲酸的安全性已成为人们关注和争论的焦点。噻二唑(2-Mercapto-5-methyl-1,3,4-thiadiazde,MMTD),学名2-巯基-5-甲基-1,3,4-噻二唑,溶于热水、甲醇、乙醇,是生产头孢菌素类等医药的重要中间体,具有抗菌性。有研究发现,MMTD对斑马鱼的胚胎具有一定的毒性[7]。
2017年第132号[8]指出严禁生产企业在小麦粉中添加噻二唑、曲酸等非食品原料。目前国内外检测方法集中在对曲酸的检测分析,如有电化学分析法[9]、近红外光谱法[10]、胶束电动毛细管色谱法[11]、高效液相色谱法[12]、液质联用测试[13]。关于食品中曲酸和噻二唑同时检测的报道较少,本文建立了UPLC-MS/MS法同时测定面粉中曲酸和噻二唑,为安全监管提供可行性的检测方法。
1 材料与方法
1.1 材料与试剂
曲酸标准品(CAS No. 501-30-4,纯度99.9%),Be
Pure公司;噻二唑(CAS No. 29490-19-5,纯度100%),
BePure公司;甲酸(HPLC级,纯度99.0%),美国DiKMA公司;乙腈(HPLC级,纯度99.9%),美国DiKMA公司;甲酸铵(AR级),上海凌峰化学试剂有限公司;实验用水均为一级水,符合GB/T 6682-2008要求。
1.2 仪器与设备
ACQUITY UPLC H-Class系列超高效液相-串联Waters XEVO TQ-S micro型三重四级杆串联质谱仪(配有电喷雾离子源(ESI)及MassLynx V4.2 SCN 1001数据处理软件),美国Waters公司;SF-TGL-16M型高速离心机,上海菲恰尔分析仪器有限公司;XW-80A
型漩涡器,上海青浦沪西仪器厂;950DAE超声仪,美国ETL TESTING公司;JA2003A型电子天平,上海精天电子仪器有限公司;Milli-Q型超纯水仪,密里博中国有限公司;0.45 ?m针式聚四氟乙烯滤膜,天津市津腾实验设备有限公司;100 ?L、1000 ?L移液枪,德国Eppendorf公司。
1.3 方法
1.3.1 色谱条件
色谱柱:Waters ACQUITY UPLC? BEH C18
(2.1 mm×100 mm,1.7 ?m),柱温:45 °C,流速:
0.45 mL·min-1,进样量为10 ?L,梯度洗脱,程序见表1。
1.3.2 质谱条件
采用多反应监测(MRM);脱溶剂温度:500 ℃,脱溶剂气流速:1 000 L/H,毛细管电压:3.0 kV,离子源温度:150 ℃,孔电压15 V,驻留时间:0.075 s。采用正负模式切换的电喷雾离子源,曲酸电喷雾为正离子模式(ESI+),定量离子对143.1/69,碰撞能为15 v;定性离子对143.1/97.1,碰撞能15 v。噻二唑电喷雾为负离子模式(ESI-),定量离子对130.8/57.9,碰撞能15 v;定性离子对130.8/89.8,碰撞能5 v。
1.3.3 前处理方法
称取混匀后的样品1.00 g,加入40 mL纯水,超声提取30 min,离心后,取上清液过0.45 ?m滤膜,注入液相色谱串联质谱仪测试。
2 结果与分析
2.1 质谱条件的选择
一般正离子模式适合于碱性样品,负离子模式适合于酸性样品。分别配制浓度为1.00 mg·L-1的曲酸和噻二唑单标标准溶液在不接色谱柱的条件下,使用电喷雾正离子模式(ESI+)进行曲酸母离子全扫描和电喷雾负离子模式(ESI-)进行噻二唑母离子全扫描。确定曲酸的母离子m/z 143.1和噻二唑的母离子m/z 130.8,利用手动优化模式对它们的子离子、孔电压和碰撞能等参数进行优化,确认优化后的参数,根据优化后的最佳质谱条件建立曲酸的ESI(+)和噻二唑ESI(-)的多反应监测方法。
2.2 色谱柱的选择
比較了Waters CORTECS? HILIC和Waters ACQUITY
UPLC? BEH C18两种柱子。经实验表明,200 ng的噻二唑在采用Waters CORTECS? HILIC柱上响应值高,但对曲酸的保留性能很差,而选择Waters ACQUITY UPLC? BEH C18 作为分离柱,曲酸(图1中1)和噻二唑(图1中2)可以同时在色谱柱上有保留(图1所示),能完全分开。
2.3 前处理提取液的选择
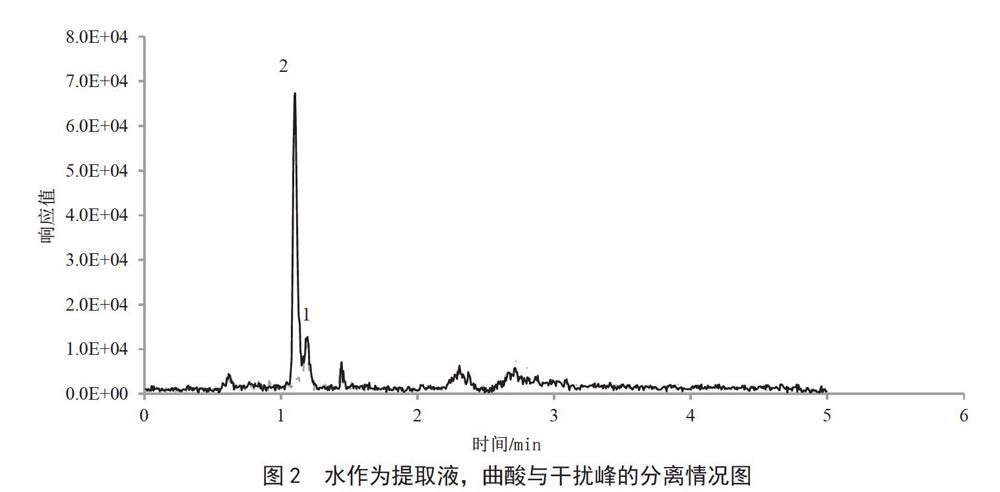
根据曲酸和噻二唑都易溶于水的性质,考察了水、乙腈和50%乙腈水作为提取溶剂的提取效率。在阴性样品进行标准溶液的添加,曲酸的添加量为0.1 mg·kg-1,噻二唑的添加量为1.0 mg·kg-1。实验表明,含有乙腈的提取溶剂,对噻二唑的提取效率没有影响,但在曲酸出锋位置有个干扰峰,没法分开,影响了曲酸的定性及定量,而水作为提取溶剂时,干扰峰(图2中的1)可以与目标峰(图2中的2)分开,如图2所示,同时也满足了提取的回收率要求,所以选择水作为提取溶剂。
2.4 线性范围和方法的检出限、定量限
将配有曲酸1.00、2.50、5.00、10.00、25.00 ?g·L-1和50.00 ?g·L-1,噻二唑10.00、25.00、50.00、100.00、250.00 ?g·L-1和500.00 ?g·L-1的混合系列基质标准曲线溶液注入超高效液相色谱串联质谱仪测试,分别以峰面积对曲酸和噻二唑的质量浓度进行线性回归,绘制标准曲线。
参考GB/T 27417-2017《合格评定 化学分析方法确认和验证指南》中空白偏差法评估方法检出限和定量限。具体操作如下:分别在10个空白样品中加入曲酸和噻二唑混合标准溶液,制成含有曲酸2.50 ?g·L-1和噻二唑25.00 ?g·L-1的样品溶液,分别注入超高效液相色谱串联质谱仪测试,得曲酸和噻二唑含量,分别计算曲酸和噻二唑含量的3倍标准偏差即为方法检出限,10倍标准偏差即为方法定量限。结果表明,曲酸在1.00~50.00 ?g·L-1,噻二唑在10.00~500.00 ?g·L-1时,标准曲线的相关系数r大于0.995,方法的检出限和定量限如表2所示。
2.5 准确度和精密度
对阴性面粉样品进行曲酸和噻二唑标准溶液的加标回收实验,曲酸的加标水平分别为0.10、0.20和0.40 mg·kg-1,噻二唑分别为1.00、2.00和
4.00 mg·kg-1,采用1.3.3的前处理方法对样品进行处理,每个添加水平重复测定6次,计算曲酸和噻二唑在面粉样品中的加标回收率及相对标准偏差。由表3可知,曲酸的回收率为100.00%~105.00%,相对标准偏差RSD为2.50%~10.72%,噻二唑的回收率为86.00%~89.00%,相对标准偏差RSD为3.65%~9.97%,方法的加标回收和精密度满足质量分析的要求
