基于分子对接技术天然药物挥发油抗抑郁研究*
2020-10-18李远洋李玉姗王芳芝余耀琨曾靖雯
李远洋,李玉姗,王芳芝,余耀琨,曾靖雯
(肇庆学院食品与制药工程学院,广东 肇庆 526061)
单胺氧化酶(MAO)主要分布在脊椎动物的大脑和肝脏中,单胺氧化酶有两种亚型,分别为MAO-A和MAO-B,其中MAO-A是抗抑郁药物研发的靶点之一。主要分布在儿茶酚胺能神经元中,使单胺类神经递质失活。单胺氧化酶抑制剂通过抑制单胺氧化酶的对单胺类物质的氧化活性,达到减轻或者消除由各种原因引起的单胺类神经递质减少或单胺氧化酶活性过高导致的疾病。
抑郁症是现代患病率较高的精神疾病之一,成因复杂,药物治疗仍然是目前临床最重要的治疗方式,大多数抗抑郁药都是基于增强体内单胺能神经传递浓度的基础上研发的,如三环类抗抑郁药物、单胺氧化酶抑制剂及选择性五羟色胺再摄取抑制剂,前两者药物副作用较大,从天然药物中寻找耐受性更好,更安全的药物为治疗抑郁症提供了新的方向。
许多天然药物挥发油被证实具有治疗、缓解抑郁症的效果[1-2],例如迷迭香与柠檬草挥发油处理后的小鼠表现出行为性活跃有所恢复[3],具有一定的抗抑郁作用。
本项工作中以MAO-A为靶点蛋白,应用DS分子模拟软件,运用CDOCKER对接方法,在CHARMnm力场下进行靶点蛋白MAO-A与从天然药物中选取的小分子配体的对接,研究天然药物挥发油组分与靶点蛋白MAO-A的相互作用,为抗抑郁药物苗头化合物的发现与结构修饰提供思路。
1 实 验
1.1 配体小分子的准备
根据文献报导[3-4,6-12],查得可能具有抗抑郁作用的挥发油主要成分12种:梨醇酯、芳樟醇、迷迭香酸、香叶木素、木犀草素、乙酸芳樟酯、薄荷酮、D-柠檬烯、松油醇、α-松油烯、β-细辛醚、α-细辛醚,通过DS直接构建天然药物挥发油组分结构,或者利用ChemOffice[13]画图拷贝到DS。考虑对映异构体、pH等因素,通过DS产生三维结构、加氢、产生异构体等。经过处理产生配体小分子,随后依次利用DS中的Prepare Ligands,Full Minimization功能,对配体小分子进行结构及力场优化处理,将处理后的小分子文件导出为PDB格式备用。
1.2 MAO-A受体蛋白的准备
PDB蛋白质结构数据库是目前最主要的收集生物大分子(蛋白质、核酸和糖)2.5维结构的数据库。
本研究选取MAO-A受体蛋白来自于RCSB数据库(PDBID:2Z5X),通过Discovery Studio软件对数据库下载的受体蛋白PDB文件进行去除水分子、原配体分子和蛋白分子加极性氢处理,最终得到所需MAO-A受体蛋白PDBQT文件。
1.3 对接环境准备
使用DS提取靶点蛋白MAO-A构象,去除多余氨基酸残基,并保存为pdb文件,查阅相关文献[1-8],以蛋白空腔拓展为活性中心,SBD_Site_Sphere球体参数设置与对接验证见表1,配体参数设置完成后均能与2z5x原配体肉叶芸香碱成功对接,对接结果与文献[15]同一数量级,验证参数设置有效。

表1 Discovery Studio模拟对接中蛋白活性位点设置参数Table 1 Parameters of protein active site setting in Discovery Studio docking
1.4 小分子与受体蛋白模拟对接
分子对接是通过把配体分子放在受体活性位点的位置,然后按照几何互补、能量互补以及化学环境互补的原则来实时评价配体与受体相互作用的强弱,并找到两个分子之间最佳的结合模式。其中包括CDOCKER-精准分子对接、全柔性受体-配体对接、DS LibDock-快速的分子对接以及分子对接后的构象分析,本研究中选用CDOCKER-精准对接模式[17]模拟MAO-A与天然药物挥发油成分小分子对接。
1.4.1 受体结合位点的选择
定义结合位点对分子对接结果有直接影响,定义结合位点主要有三种方法,一是配体扩张法,即以原配体(共晶化合物)所在位置为活性中心扩张一定的范围,此范围内的氨基酸残基则定义为活性位点;二是文献检索法,即通过文献检索找到已报道的活性位点来定义结合位点;三是软件预测法,即通过DS软件分析最有潜力的结合位点。
由于该蛋白有共晶化合物,因此本研究主要运用文献检索法与活性中心扩张法定义受体结合位点。设置原配体所在位置为活性中心,通过调整SBD_Site_Sphere球体半径及位置,令球体的空腔即为活性位点。原则上,要保证球体将配体全部包裹,但球体大小不宜过大,否则会使不同的小分子对接至受体的其他位置造成结果偏移,球体的大小会直接影响对接结果。
1.4.2 精准分子对接技术(CDOCKER)
采用DS中的CDOCKER对接技术进行挥发油成分小分子与MAO-A受体蛋白模拟对接,CDOCKER是在CHARMm立场下产生高精度分子对接结果的技术。通过对电子(量子论)与原子(立场法)的比较,选用适用于生物大分子的模拟,计算量相对小10倍的分子立场法。在适当范围内,该法较量子论计算方法的精准度相差无几。DS中的CHARMm力场函数复杂度较低,精确度较高。函数关系如下:
Epot=Ebond+Eangle+Etorsion+Eoop→internal terms
+Eelec+EvdW→internal/external terms
+Econstraint+Euser→special
MAO A从PDB数据库获得,快速直接将蛋白质溶解在不同的pH值下,设置显性溶剂环境,为避免原子受到较大的扰动而造成体系计算失败(特别是体系升温过程)。进行能量优化消除蛋白结构内部蛋白质与水分子之间的一些触碰及结构不合理区,同时采用PME法进行静电处理。采用缓慢升温(1ps升温10 K),即升温过程选择true,避免直接将体系温度升高到较高的体系模拟的温度时,体系内原子位移较大而计算失败的现象。待体系经过一定时间达平衡后进行采样。并从MD的采样数据中预测二面角、RMSD、体系能量、势能、氢键数量、配体受体氢键作用的变化。
首先对靶点蛋白,配体小分子进行结构及力场优化处理,其次设置好活性位点,球体的坐标和半径,Top Hits等参数后运行,进行精准分子对接。
1.4.3 分子对接构象结果分析
分子之间的非键相互作用是生命体系中分子识别的基础,药物设计的关键之一就是分析分子间的相互作用情况。DS中的对接结果报告将对接过程中的各种相互作用力总结表述,包括疏水、氢键、静电以及其他作用,明确受体蛋白中的各种氨基酸残基与配体小分子之间的相互作用的数目以及每一种相互作用类型涉及数目排行靠前的氨基酸残基。
(1)配体-蛋白相互作用的二维平面图生成
该平面图可显示受体蛋白氨基酸残基与配体对接构型间的非键相互作用类型及距离等相关信息,便于我们更直观的观察两者的相互作用及关键的氨基酸和基团,以便分析对接结果。
(2)作用类型热图生成
对接完成后打开DS中的Heat Map(Favorable)配体分子与受体蛋白之间的相互作用以热图的形式被展现出来。热图中包含了所有的对接构象及相对应的相互作用信息。
2 结果与讨论
2.1 挥发油主要成分小分子与受体蛋白结合情况
通过DS中的CDOCKER精准分子对接技术模拟12种挥发油成分小分子与MAO-A受体蛋白模拟对接,对接结果中选取最优对接构象,并对能量排序见表2。CDOCKER对接主要计算配体小分子与受体蛋白总能量(CDOCKER_ENERGY),配体-受体相互作用能量(CDOCKER_INTERACTION_ENERGY),以此两种能量做对接情况的参考,CDOCKER_ENERGY越小,说明对接体系总能量低,对接体系稳定,CDOCKER_INTERACTION_ENERGY越低,表示对接过程中相互作用小,两者结合得越好。DS中对上述两种能量数值取负数,故-CDOCKER_ENERGY越大,对接越稳定, -CDOCKER_INTERACTION_ENERGY越大,配体与受体结合越好。取两值之和并对其排序,可作为对接情况优劣的参考依据。

表2 挥发油成分小分子与MAO-A对接结果Table 2 Docking results of volatile oil components and MAO-A
挥发油主要成分小分子均能与MAO-A对接成功,且与原配体肉叶芸香碱相比较,有6种配体小分子对接结果较优于原配体。其中前3种配体与受体蛋白结合二维平面图如图1~图3所示。
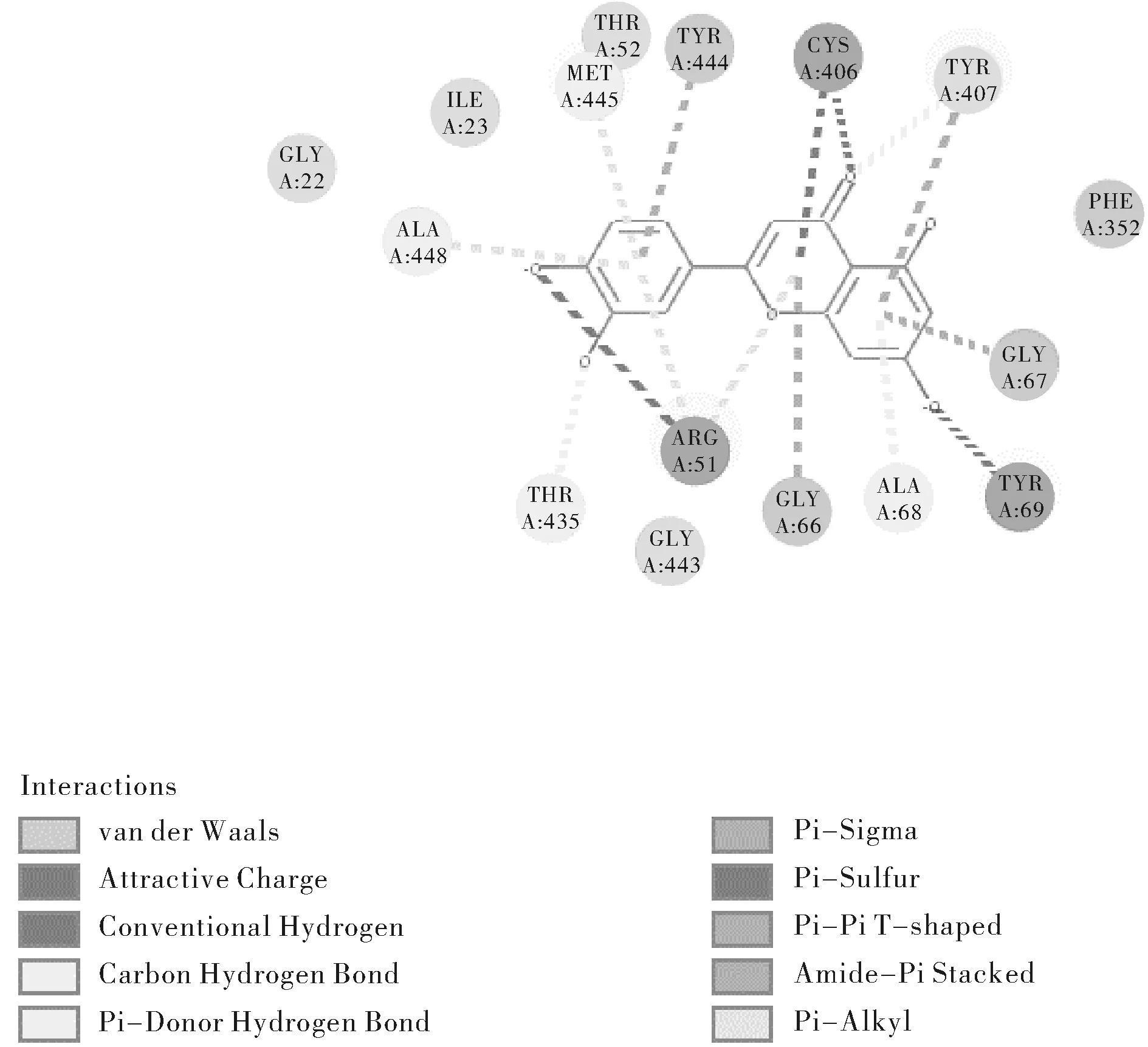
图1 木犀草素与MAO-A对接平面图Fig.1 Figure of luteolin docking with MAO-A
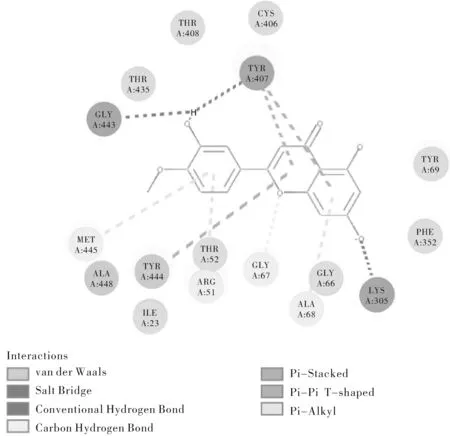
图2 香叶木素与MAO-A对接平面图Fig.2 Figure of vanillin docking with MAO-A
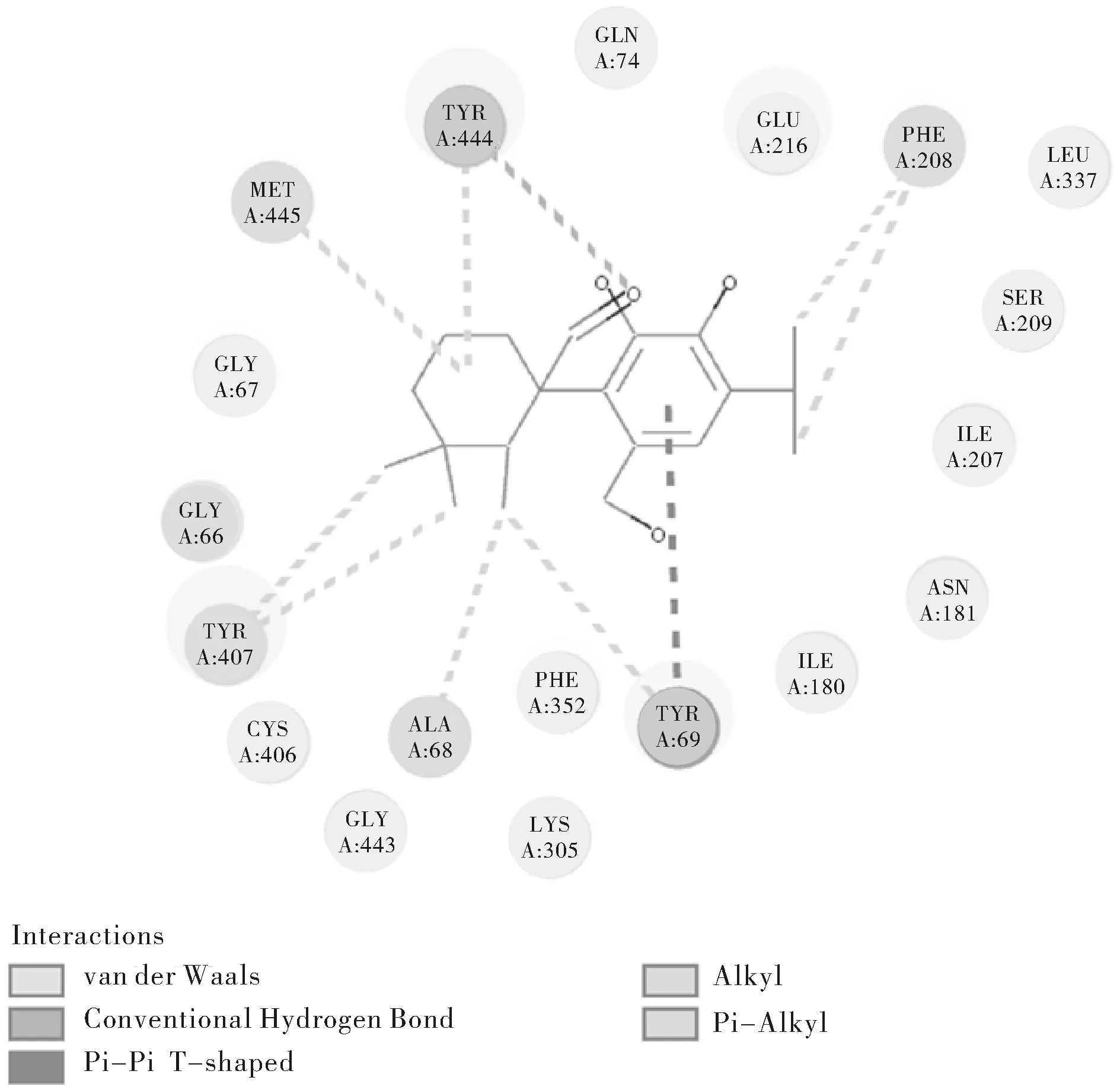
图3 迷迭香酸与MAO-A结合Fig.3 Figure of rosmarinic acid docking with MAO-A
2.2 天然药物小分子与受体的各种作用力
配体分子与受体蛋白的各种作用力统计与各种作用数量前五的残基计数见表3。
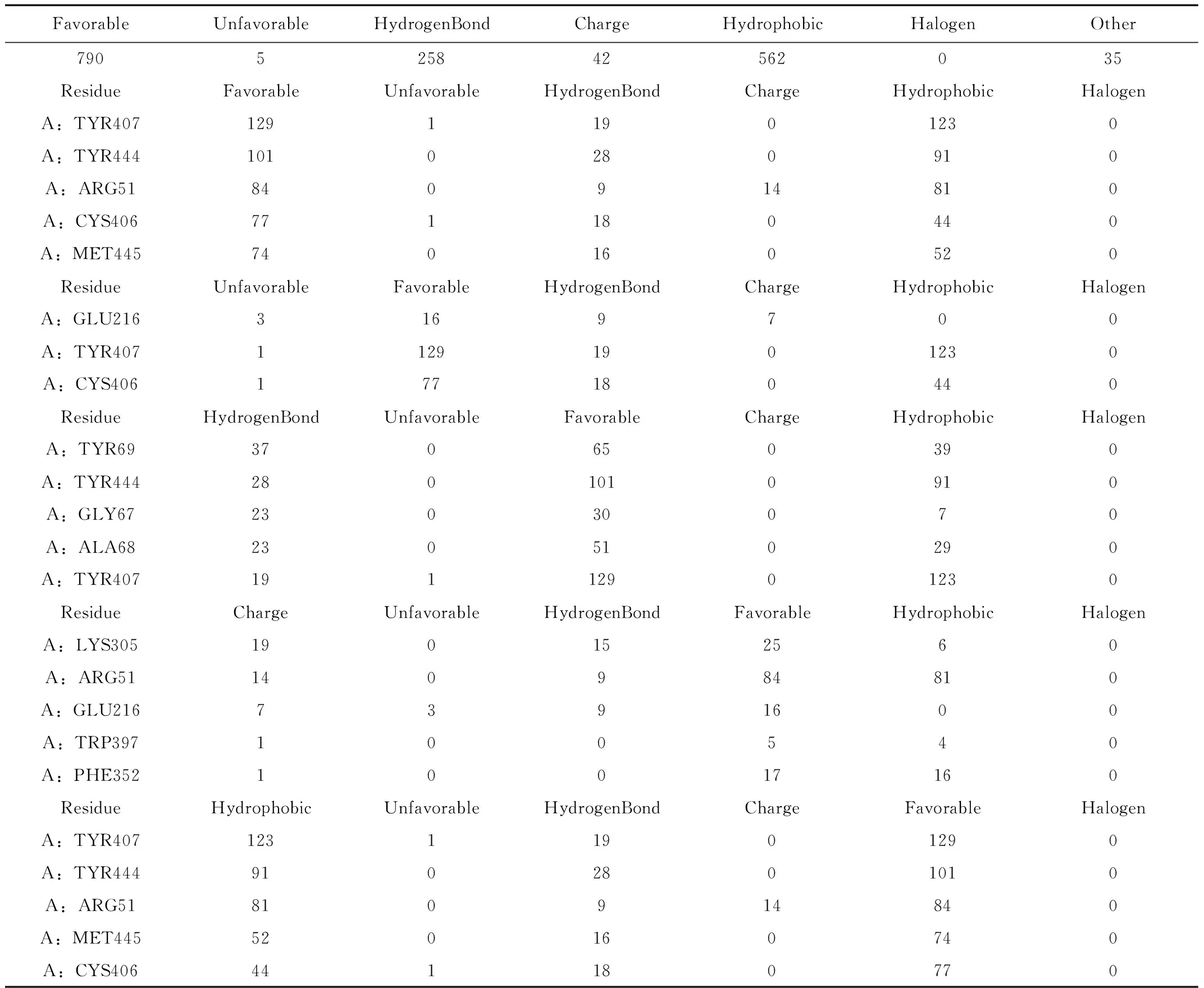
表3 全类型作用总计Table 3 Total Interaction count
对接过程中各种对接形态配体与受体蛋白中产生氢键作用的残基共有258个,产生疏水作用的有562个,静电作用的有42个,其中产生氢键数目排行前五的氨基酸残基为:TYR69、TYR444、GLY67、ALA68 、TYR407;产生静电相互作用的残基数目排行前五的氨基酸残基为:LYS305、ARG51、GLU216、TRP397、PHE352;产生疏水作用的残基数目排行前五的氨基酸残基为:TYR407、TYR444、ARG51、MET445、 CYS406。产生其他作用的残基有CYS406与MET445。
对接中全部配体与受体产生的所有作用如图4所示,可见各配体与氨基酸残基互相作用数目热度,其中互相作用热度较高的有ARG51、TYR69、GLY66、TYR407等。可知MAO-A蛋白的活性空腔大致由上述残基共同构成,且其中氨基酸残基TYR407热度较高,可知其在结合过程中是关键残基[17]。
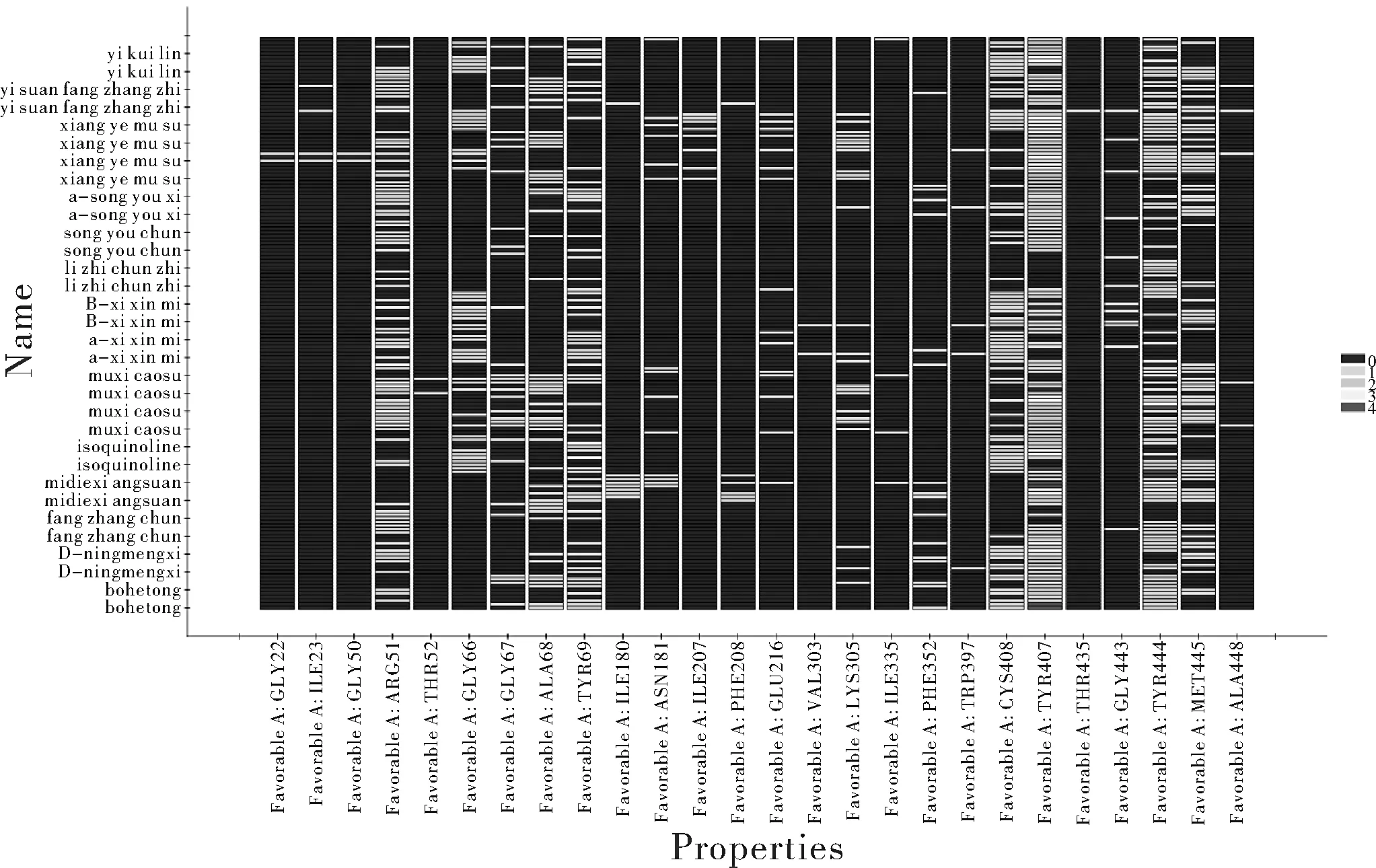
图4 配体与受体相互作用热图Fig.4 Heat map of ligand-receptor interaction
2.3 数据评判标准
除DS中的对接结果中的能量数据之外,研究结果[17]表明MAO-A的活性中心结合的关键在于与氨基酸残基Phe208、ASn181、Tyr407产生相互作用,而本研究选用的MAO-A(PDBID:2z5x)的原配体肉叶芸香碱与氨基酸残基Tyr407形成π-π堆积作用,也从一定程度说明了配体小分子与上述残基的相互作用是配体小分子与受体蛋白活性中心结合的关键,故可从配体分子与残基的作用情况与结合的数据结果分析配体小分子是否能与受体蛋白活性中心结合。根据结合作用热图可知,TYR407热度较其他残基更高,表明对接结果数据可靠。
3 结 论
本研究从已知的天然药物挥发油成分中选择可能具有抗抑郁成分12种,与受体蛋白原配体肉叶芸香碱对比结果分析,共有6种对接结果得分高于原配体,天然药物挥发油活性小分子作为抑制剂进入受体“口袋”形成空间位阻效应,最终抑制MAO-A,阻止脑内5-HT和NE降解,同时提升脑内突触间隙5-HT和NE的浓度,起到一定的抗抑郁作用。
以本次对接结果中能量高低排序,得分较高的3种小分子为例:
木犀草素与MAO-A中的TYR407氨基酸残基可产生T型的π-π堆积作用(Pi-Pi T-shaped),与GLY66、TYR44、TYR69等氨基酸残基构成的活性空腔相结合;
香叶木素环状结构与MAO-A中的TYR407、TYR444产生π-π堆积作用,酚羟基与LYS305起Salt Bridge作用;
迷迭香酸中的环状结构与残基TYR69产生T型π-π堆积作用(Pi-Pi T-shaped),两个甲基与TYR407产生Pi-Alkyl作用。
就以上三种对接能量得分较高的配体小分子对接情况而言,均能与TYR407残基产生π-π堆积作用,且能与关键氨基酸残基构成的活性空腔良性结合,可能是其对接能量得分较高的原因之一。
研究中选用的12种小分子多数存在于天然药物挥发油中,且来源并不单一,对接得分较高的几种配体在临床中抗抑郁活性有待进一步活体实验验证,对接得分不高的配体也可能通过其他途径起抗抑郁作用,同样能起到抗抑郁作用的受体蛋白还有多巴胺转运体(DAT)、5-羟色胺转运体(SERT)等,抗抑郁作用机制并非单一渠道,与MAO-A作用只是其中一种抗抑郁途径,计算机模拟分子对接结果可为天然药物挥发油抗抑郁研究提供理论导向,为抑郁症新药研发提供新的思路。
