F原子对过渡金属铱配合物结构和光谱性质的影响
2020-10-18聂建航王子轩王天奇张建坡
聂建航,王子轩,王天奇,张建坡
(吉林化工学院 化学与制药工程学院,吉林 吉林 132022)
近几十年来,有机发光二极管(OLED)因低功耗和有可能以适中的价格制造出大而明亮的平板显示器,在墙壁照明和微型显示屏等方面有着广泛的应用.特别是基于2-苯基吡啶的铱配合物衍生产品引起了更多的关注,它们可以通过改性ppy配体和引入不同的辅助配体而发出多种颜色的光[1-2].例如,含氟苯基吡啶配体Ir(III)配合物发光性能良好,陆续报道合成了[Ir(Fnppy)3]、[(Fnppy)2Ir(acac)][n=3,F3ppy=2-(3,4,6-三氟苯基)吡啶;n=4,F4ppy=2-(3,4,5,6-四氟苯基)吡啶;acac=乙酰丙酮][3-5]配合物,研究表明引入吸电子基团使部分C—H键和C—F键活化,从而减少辐照激子衰变,提高光致发光(PL)效率和电子迁移率,且在苯环上取代的氟原子的位置和数量主要影响铱配合物的光谱性质.为了探究F原子对过渡金属铱配合物结构和光谱性质的影响,本文采用量子化学计算方法,选取一类(C^N)2Ir(idpt)配合物[6]进行了详细的理论研究,阐明了此类配合物的发光规律,揭示了F原子在分子结构和光谱性质中的具体作用.
1 计算方法
3个配合物在优化中均无对称性约束.使用密度泛函方法中的B3LYP[7]和UB3LYP[8]泛函分别对其基态和激发态几何结构进行全优化.在基态、激发态几何结构的基础上,使用含时密度泛函(TD-DFT)[9]方法联合CPCM溶剂模型计算了配合物在CH2Cl2溶剂中的光谱特征.由于idpt为辅助配体,配体上的苯基取代并不会对配合物的发光颜色和结构造成重要的影响,因此为了节约计算成本,优化中不包含苯基,这种计算方法也被证实了是准确、可靠的[10].基组选择如下,Ir原子使用LanL2DZ基组[11],计算中只考虑了其最外层的价电子,C、N、H、O、P和F原子使用6-31G*基组.以上计算通过执行Gaussian09(E02)程序包[12]在大型曙光服务器上完成.
2 结果与讨论
2.1 F取代对分子几何结构的影响
图1展示出配合物1-3的几何结构及代表性原子编号,其基态和激发态结构参数及配合物3的实验值列在表1.如表所示,基态时3的Ir-N(1)(2.060 Å)、Ir-C(1)(2.005 Å)、Ir-O(1)(2.247 Å)分别与其实验值相了0.005 Å、0.01 Å和0.049 Å,此误差在实验值和理论计算允许范围内,说明所计算的分子结构合理、准确.对比3个分子的键长、键角和二面角,F原子的引入并没有使其主体结构发生较大改变,分子2和3与分子1相比,所有数值都非常接近,只有二面角O1-P1-N3-P2变化略大,表明F原子加入越多,导致idpt配体的扭曲程度就越大,但基本不影响C^N配体的刚性结构.二面角C1-Ir-N1-O1、C1-Ir-N1-C2均接近于90°,表明3个双齿配体近乎垂直,配合物呈类似八面体的稳定结构.

图1 配合物1-3的优化几何结构
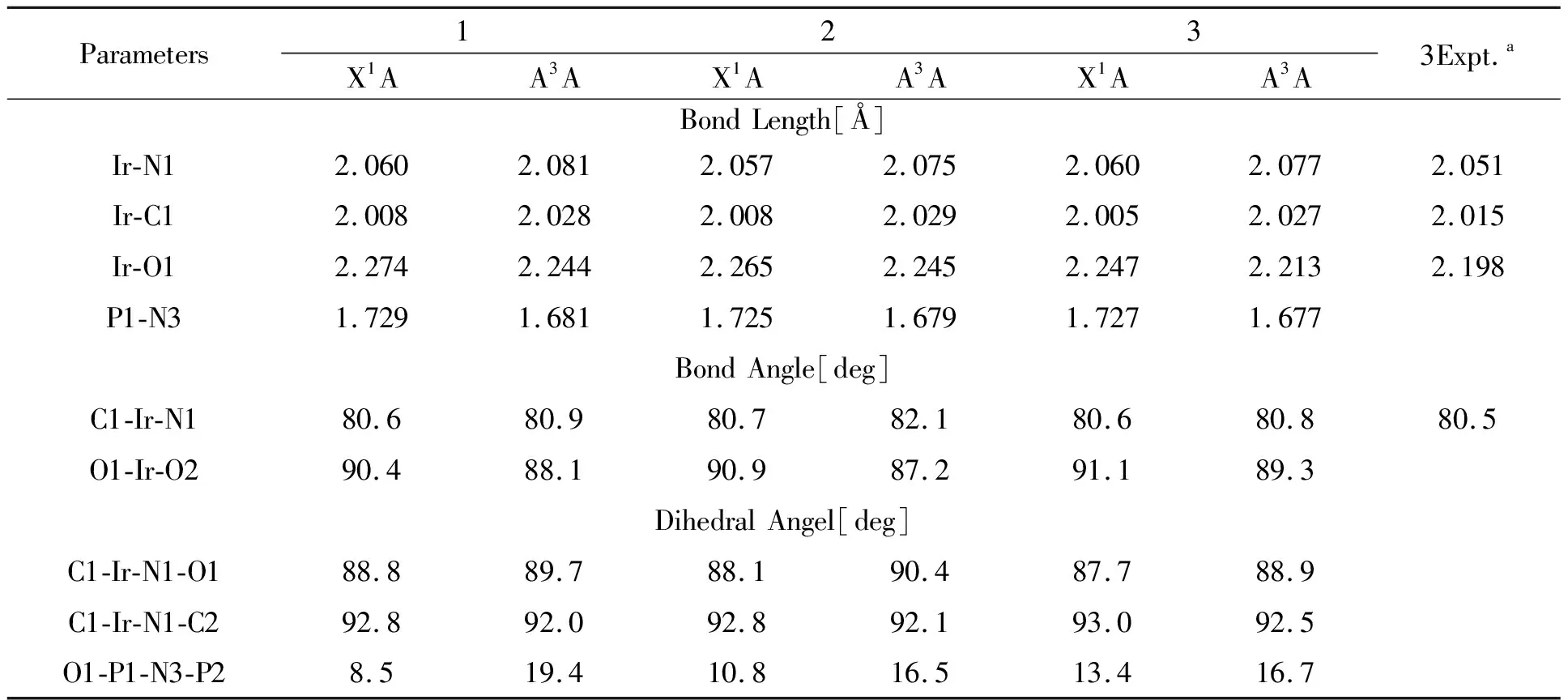
表1 配合物1-3的基态和三重激发态几何结构参数及其实验值
激发态与基态相比,三个分子的Ir-N1、Ir-C1变长,而Ir-O1、P1-N3变短,这归因于基态时刚性C^N配体和铱阳离子具有强的π堆积相互作用,在激发态时金属的电子云向idpt配体转移,削弱了金属和C^N配体之间的相互作用,增强了idpt配体本身的电子云排布,对应着最低能吸收和发射的MLCT跃迁本质.同样激发态时二面角O1-P1-N3-P2变化较明显,idpt配体发生了不同程度的扭曲,与分子1相比,激发态时F原子的加入反而适当的抑制了idpt配体的扭曲.
2.2 F取代对分子吸收光谱的影响
在基态结构基础上,利用含时密度泛函方法得到了1-3在CH2Cl2溶剂中的吸收光谱.表2给出了选择的代表性吸收的光谱数据,包括跃迁轨道、振荡强度、能量和跃迁性质.其涉及到的跃迁分子轨道成分展示在表3.用Gaussian型曲线拟合的吸收光谱展示在图2.

表3 1-3在B3LYP泛函水平下的基态分子轨道成分

Wavelength/nm
由图2可知,此类配合物具有3个典型的吸收带.来自于表2,计算得到1-3的最低能吸收分别在447(2.77)、452(2.74)和424 nm.配合物1的447 nm的吸收来自于分子轨道118→119的激发,分子轨道118是最高占据分子轨道(HOMO),由53.1%的d(Ir)、32.1%的(C^N)和14.8%的(idpt)构成,而119是最低空轨道(LUMO),主要由66.6%的(C^N)和27.3%的(idpt)配体占据.因此,该跃迁是属于d(Ir)+(C^N)+(idpt)→*(C^N)+*(idpt)的金属到配体以及发生在N^N和idpt配体之间的电荷转移跃迁(MLCT/LLCT).配合物2-3的最低能吸收也都来自HOMO→LUMO的跃迁,并与1具有相似的跃迁性质,其区别在于由于F原子的引入,LUMO轨道上的C^N配体成分显著增加到80%以上,导致idpt配体参与的程度下降.分析3个分子的最低能吸收波长可发现,少量的F原子引入并没有造成波长的显著蓝移,甚至有小量的红移趋势,但是随着F原子数量的增加,可使波长出现明显的蓝移.
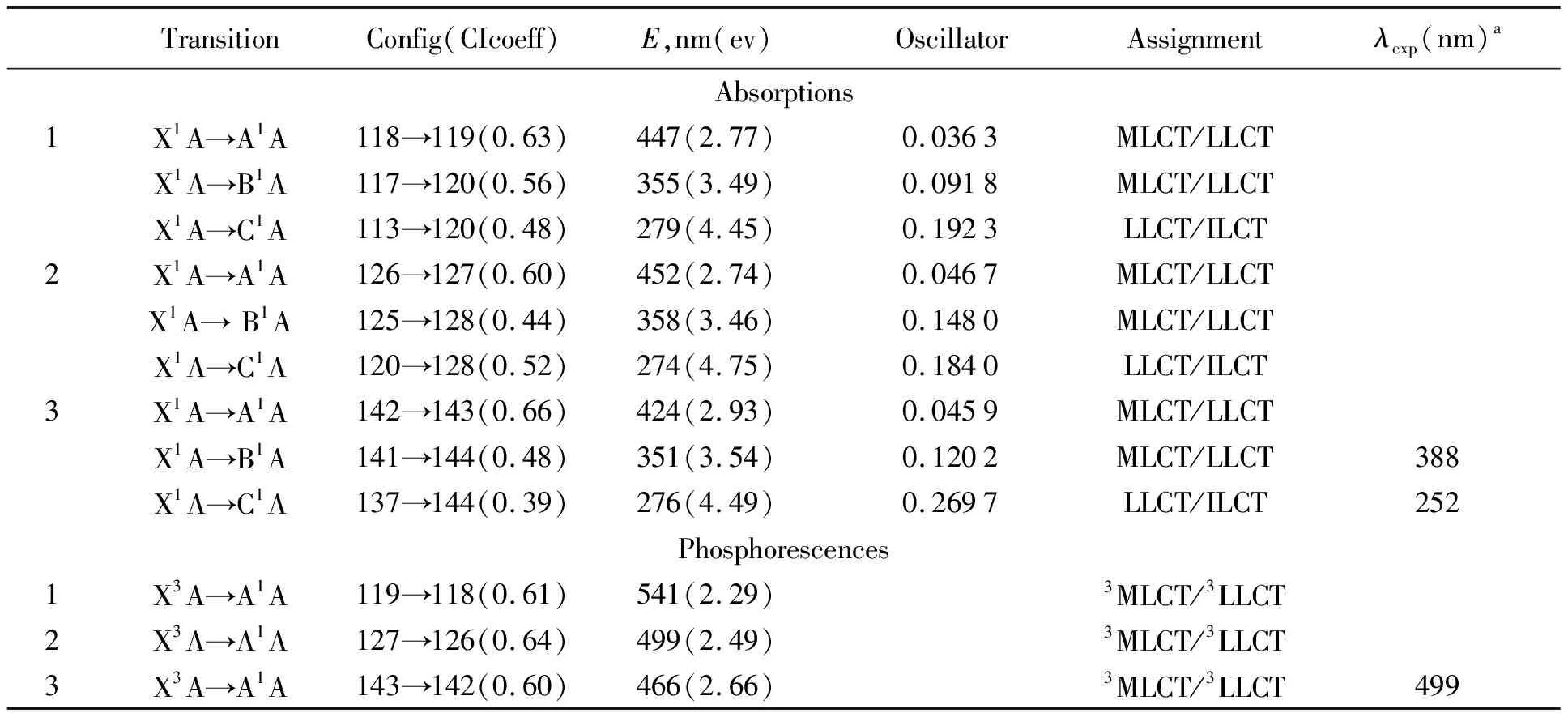
表2 1-3在CH2Cl2溶剂中的吸收和发射光谱数据及其3的实验值
考虑第二个吸收带,与低能吸收相比它们的振荡强度有了明显提升,跃迁波长都集中在350~360 nm之间,波长的差异非常小.依据上边的跃迁分析,它们都来自于HOMO-1→LUMO+1的跃迁,该跃迁的起始轨道和终止轨道占据情况与最低能吸收类似,因此也具MLCT/LLCT跃迁性质.分析该处的吸收波长可发现,F原子的引入并没有对此处跃迁产生明显影响.
对于第三个吸收带,从图2可看到,该处吸收具有最大的震荡强度,跃迁波长也非常相近在270~280 nm之间.来自于分子1,279 nm的吸收属于分子轨道113→120的激发捐献,分子轨道113由59.3%的(idpt)和37.8%的(C^N)配体占据,而120则主要由C^N配体捐献.因此,该跃迁拥有LLCT/ILCT混合跃迁特征.分子2和3也拥有类似的跃迁性质.其区别在于,分子3的跃迁起始轨道有83.6%的(idpt)成分,因此该跃迁可归属于LLCT跃迁,ILCT跃迁比例可近似忽略.
2.3 F取代对分子发射光谱的影响
配合物1-3在CH2Cl2溶液中的磷光光谱数据如表2所示.三个配合物的最低能磷光发射分别在541 nm(1)、499 nm(2)和466 nm(3).与其最低能吸收相类似,它们都被指认为起源于HOMO →LUMO的激发,HOMO轨道主要占据在金属和两个双齿配体上,而LUMO轨道由两个双齿配体捐献,因此与最低能吸收具有相似的跃迁性质.比较1-3的最低吸收能和发射(HOMO→LUMO),它们都具有相似的轨道成分和跃迁性质,其能量差分别为0.48 eV、0.25 eV和0.27 eV,F原子的引入明显导致其斯托克斯频移变小.比较三个分子的发射波长也能发现,吸电子基团的增加会导致其波长明显的蓝移,甚至可以通过调整F原子的数量来改变此类配合物的发光颜色.
3 结 论
本文对3个含有苯基吡啶、亚氨基二磷酸盐配体铱配合物进行了详细地理论研究.通过改变苯基吡啶配体上F原子的数量探究了对其结构和光谱性质的影响.研究结果表明:F原子的加入对配合物的主体结构影响不大,但是可以导致亚氨基二磷酸盐配体发生明显地扭曲.F原子通常会降低HOMO和LUMO的轨道能量,但由于HOMO轨道能量降低的更多,使HOMO-LUMO能级间隙变大,从而导致发射波长蓝移.因此,此类分子可以通过调节F原子的数量来改变配合物的发光颜色,从而有望实现黄绿光到紫光的全色显示.
