一测多评法测定复方金线莲中4种活性成分的含量
2020-10-15许晓文洪瑞霞杨丽娟黄丽英
许晓文, 洪瑞霞, 杨丽娟, 黄丽英
复方金线莲口服液是由金线莲、灵芝提取液制成,具有清热解毒、益气凉血的功效,适用于急性或慢性肝炎、乙肝病毒携带者之热毒气虚证,是福建医科大学孟超肝胆医院的院内制剂。金线莲为兰科开唇兰属植物的新鲜或干燥全草,主产于福建、浙江、台湾、云南等地,具有多种药理功效,在东南亚及我国民间广泛用于治疗肝炎、高血压病、肿瘤、肾炎和糖尿病等,特别是保肝作用倍受人们青睐,在福建、广东和台湾民间广泛用于治疗肝炎[1-2]。灵芝在我国已有2 000多年的药用历史,被历代医药学家视为滋补强壮、扶正固本的神奇珍品。核苷类物质是金线莲的主要活性成分[3],具有明显的药理活性,对于急、慢性肝炎和肝硬化等具有良好的疗效[4]。三萜类化合物是灵芝中的主要有效成分(如灵芝酸A[5-6]和赤芝酸C[7]),对多种理化及生物因素引起的肝损伤有保护作用[8],能促进肝脏对药物及毒物的代谢,对于中毒性肝炎有确切的疗效,可明显消除慢性肝炎所引起的头晕、乏力、恶心、肝区不适等症状,并且能够有效地改善肝功能[9]。
如果以传统测定方法同时测定中药材中几种成分,不但检验周期长,还耗能、耗时、耗对照品。一测多评法作为一种对多指标质量控制的有效方法[10-11],不仅可测定某个代表性成分,还可计算出其他待测成分的含量,是一种适合中药特点的多指标质量评价的新模式[12-14]。鸟苷相对其他对照品价格较便宜,且含量稳定,故本研究以鸟苷对照品作为参照物,对其与尿苷、赤芝酸C、灵芝酸A的相对校正因子进行含量计算,以验证一测多评法在复方金线莲口服液多指标质量控制中应用的适用性及可行性。
1 材料与仪器
1.1仪器 液相色谱质谱联用仪(LCMS-2020型)、高效液相色谱仪(LC-15C型,SPD-15C紫外-可见检测器)及紫外可见分光光谱仪(2450型)(日本岛津公司);Agilent ZOBAX SB-Aq色谱柱(4.6 mm×250 mm,5 μm)(美国安捷伦公司);分析天平(BS124S,上海奕宇电子科技有限公司);精密pH计(UltraBasic-7,美国丹佛仪器有限公司);自动双重纯水蒸馏器(RE-2000,上海亚荣生化仪器厂);数控超声波清洗器(KQ-250DE型,昆山市超声仪器有限公司)。
1.2试剂 尿苷(纯度≥99%,批号:L1628034,Aladdin试剂公司);鸟苷(纯度≥98%,批号:E1605104,Alfa Aesar公司);赤芝酸C(纯度≥98%,批号:M11M10S82676)、灵芝酸A(纯度≥98%,批号:6032)购于Tansoole公司;色谱级的甲醇、乙腈(美国Sigma-Aldrich试剂公司);其余试剂为分析纯,均购于国药集团化学试剂有限公司;实验用水为实验室自制双蒸水。
1.3复方金线莲口服液 复方金线莲口服液(每支10 mL,生产批号:20181104,20190603,20191102,由福建医科大学孟超肝胆医院提供)。
1.4方法
1.4.1色谱条件 色谱柱:Agilent ZOBAX SB-Aq色谱柱(4.6 mm×250 mm,5 μm);流动相包括13 mmol/L乙酸铵水溶液和乙腈,二元梯度洗脱,洗脱程序见表1;流速:1.0 mL/min;检测波长:254 nm;柱温:30 ℃;进样量:20 μL。
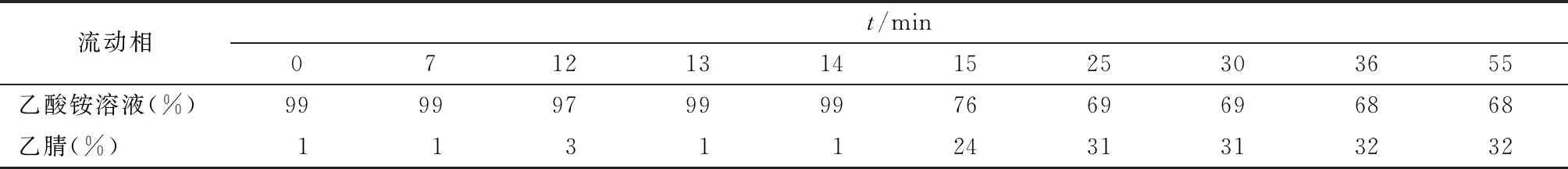
表1 流动相洗脱程序
1.4.2质谱条件 电喷雾离子源(electrospray ionization,ESI),扫描模式正负离子检测ESI(±);雾化器流速:1.5 L/min,干燥气流速(N2):12 L/min;扫描范围:质荷比150~700。接口温度:350 ℃,加热块温度:200 ℃,DL温度:250 ℃,进样量:20 μL。
1.4.3对照品溶液的制备 分别精密称取尿苷10.0 mg、鸟苷10.0 mg、赤芝酸C 9.0 mg和灵芝酸A 10.0 mg置于10 mL容量瓶中,加甲醇溶解并定容至刻度,配制成对照品溶液的浓度分别为尿苷1 g/L、鸟苷 1 g/L、赤芝酸C 0.9 g/L、灵芝酸A 1 g/L。置于4 ℃的冰箱内,待用。
1.4.4混合对照品溶液的制备 分别精密吸取对照品溶液尿苷600 μL、鸟苷600 μL、赤芝酸C 222 μL、灵芝酸A 1 600 μL,置于10 mL容量瓶中,加甲醇溶解并定容至刻度,配制成尿苷0.06 g/L、鸟苷0.06 g/L、赤芝酸C 0.02 g/L、灵芝酸A 0.16 g/L。
1.4.5阴性对照溶液的制备 按照复方金线莲口服液的处方比例和生产工艺,分别制备不含金线莲和灵芝的阴性对照溶液。
1.4.6标准曲线的制备 分别取上述混合对照品溶液8,5,2.5,1,0.5,0.25,0.062 5 mL置于10 mL容量瓶中,加甲醇溶解并定容至刻度,稀释得到7个不同浓度的混合对照品溶液。经0.22 μm微孔滤膜过滤,吸取上述混合标准系列溶液,进样20 μL,测定。
1.4.7质谱定性分析 按1.4.2项下质谱条件,对供试品溶液进行质谱定性分析。
1.4.8外标法 将同一批次的10瓶复方金线莲口服液均匀混合在一起,0.45 μm微孔滤膜滤过,即为供试品溶液。按1.4.1项下色谱条件对供试品溶液进样20 μL测定,以外标标准曲线法计算含量。测定重复3次。
1.4.9一测多评法 混合对照品溶液(尿苷30 μg/mL、鸟苷30 μg/mL、赤芝酸C 10 μg/mL、灵芝酸A 80 μg/mL)进样2,5,10,15,20 μL,分别进行测定,计算得到各自的校正因子f(f=W/A,W是物质的含量,A是检测器响应值)以及相对校正因子fkm(fkm=fk/fm,k是参照物,m是其他成分)。按1.4.1项下色谱条件对供试品溶液进样20 μL测定,获得各个成分的峰面积,以一测多评法(Wm=fkm×Ak×Wk/Ak)计算含量。上述进样测定重复3次[15]。
2 结 果
2.1HPLC-ESI/MS鉴定4种有效成分
2.1.1对照品溶液和供试品溶液的HPLC-UV分析 在1.4.1项色谱条件下,对照品溶液和供试品溶液经HPLC-UV分析得色谱图(图1),对照品中的4种有效成分尿苷、鸟苷、赤芝酸C和灵芝酸A的保留时间分别为7.23,17.29,36.37和49.17 min,各对照品的浓度分别为30,30,10和80 μg/mL。根据保留时间,复方金线莲口服液色谱图的1,2,3,4号色谱峰分别初步鉴定为尿苷、鸟苷、赤芝酸C、灵芝酸A。
2.1.2方法专属性 取阴性对照溶液,在1.4.1项色谱条件下进行分析,结果表明,金线莲汁与灵芝原液中都含有尿苷和鸟苷,且金线莲汁中不含赤芝酸C和灵芝酸A。HPLC色谱图见图1。
2.1.3对照品溶液和供试品溶液的MS定性分析 由于中药材样品中有效成分复杂,微量成分的色谱峰有干扰峰的影响,因此,对复方金线莲口服液样品进行质谱分析。在1.4.2项质谱条件下,对对照品溶液和复方金线莲口服液供试品溶液4个色谱峰按出峰顺序进行质谱分析,得到各离子峰的精确质量数,质谱图见图2。图2(A,A1)分别是对照品和样品1号色谱峰对应的质谱图,质荷比245,267分别是尿苷生成的[M+H]+准分子离子峰和[M+Na]+离子峰;图2(B,B1)分别是对照品和样品2号色谱峰对应的质谱图,质荷比284是鸟苷生成的[M+H]+准分子离子峰;图2(C,C1)分别是对照品和样品3号色谱峰对应的质谱图,质荷比475是赤芝酸C生成的[M-H]-准分子离子峰;图2(D,D1)分别是对照品和样品4号色谱峰对应的质谱图,质荷比515是灵芝酸A生成的[M-H]-准分子离子峰。根据色谱的保留时间、质谱数据和文献资料,样品溶液中色谱图峰1、峰2、峰3和峰4分别是尿苷、鸟苷、赤芝酸C和灵芝酸A。
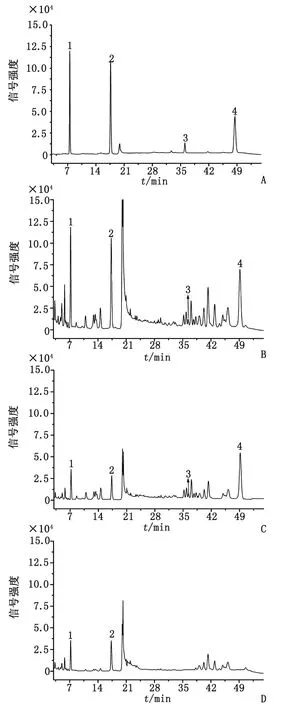
1:尿苷;2:鸟苷;3:赤芝酸C;4:灵芝酸A. A:混合对照品(尿苷和鸟苷浓度30 μg/mL,赤芝酸C 10 μg/mL,灵芝酸A 80 μg/mL); B:复方金线莲口服液; C:不含金线莲的阴性对照溶液; D:不含灵芝的阴性对照溶液.图1 混合对照品、样品和阴性对照溶液的色谱图Fig 1 HPLC choromatograms of mixture references ingredients and samples
2.2线性关系的考察、检测限和定量限 取上述8个不同浓度混合对照品溶液按1.4.1项色谱条件进行分析,以各对照品峰面积(Y)为纵坐标,各对照品浓度(X,μg/mL)为横坐标,绘制标准曲线,结果见表2,表明各化合物在相应的浓度线性范围内线性关系良好。
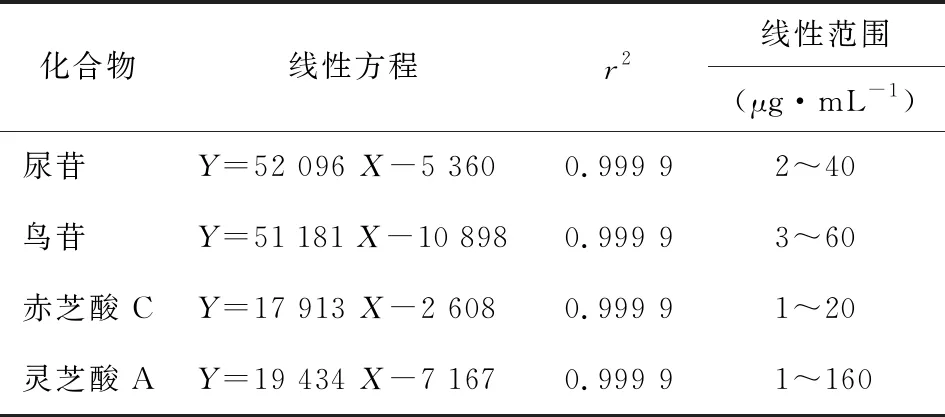
表2 线性回归方程及相关系数
2.3精密度试验 选取一定浓度的混合对照品溶液(尿苷30 μg/mL、鸟苷30 μg/mL、赤芝酸C 10 μg/mL、灵芝酸A 80 μg/mL),按照1.4.1项色谱条件连续进样6次,测定尿苷、鸟苷、赤芝酸C和灵芝酸A的含量并计算RSD值,结果分别为0.52%,1.34%,1.23%和1.17%,结果表明仪器精密度良好。
2.4重复性试验 选取一定浓度的混合对照品溶液(尿苷30 μg/mL、鸟苷30 μg/mL、赤芝酸C 10 μg/mL、灵芝酸A 80 μg/mL),按照1.4.1项色谱条件连续测定3 d,测定尿苷、鸟苷、赤芝酸C和灵芝酸A的含量并计算RSD值,结果分别为1.85%,1.64%,1.74%和2.27%,表明该方法重复性良好。
2.5稳定性试验 取复方金线莲口服液,在室温下放置0,1,2,4,6,8,12 h后各精密吸取20 μL进样,尿苷、鸟苷、赤芝酸C和灵芝酸A的色谱峰面积的RSD(n=6)分别为2.78%,2.81%,1.59%和3.17%,表明供试品在12 h内稳定性良好。
2.6加样回收率试验 精密吸取复方金线莲口服液(生产批号:20190603)0.5 mL,按低、中、高水平分别加入适量的混合对照品溶液并定容至1 mL,用0.45 μm微含孔滤膜滤过,进样20 μL,按照外标法计算各成分的含量,并计算回收率,结果表明该方法的准确度良好(表3)。

A,A1:尿苷; B,B1:鸟苷; C,C1:赤芝酸C; D,D1:灵芝酸A.图2 4种活性成分的对照品和样品质谱图Fig 2 Full mass spectrum of four standards and samples
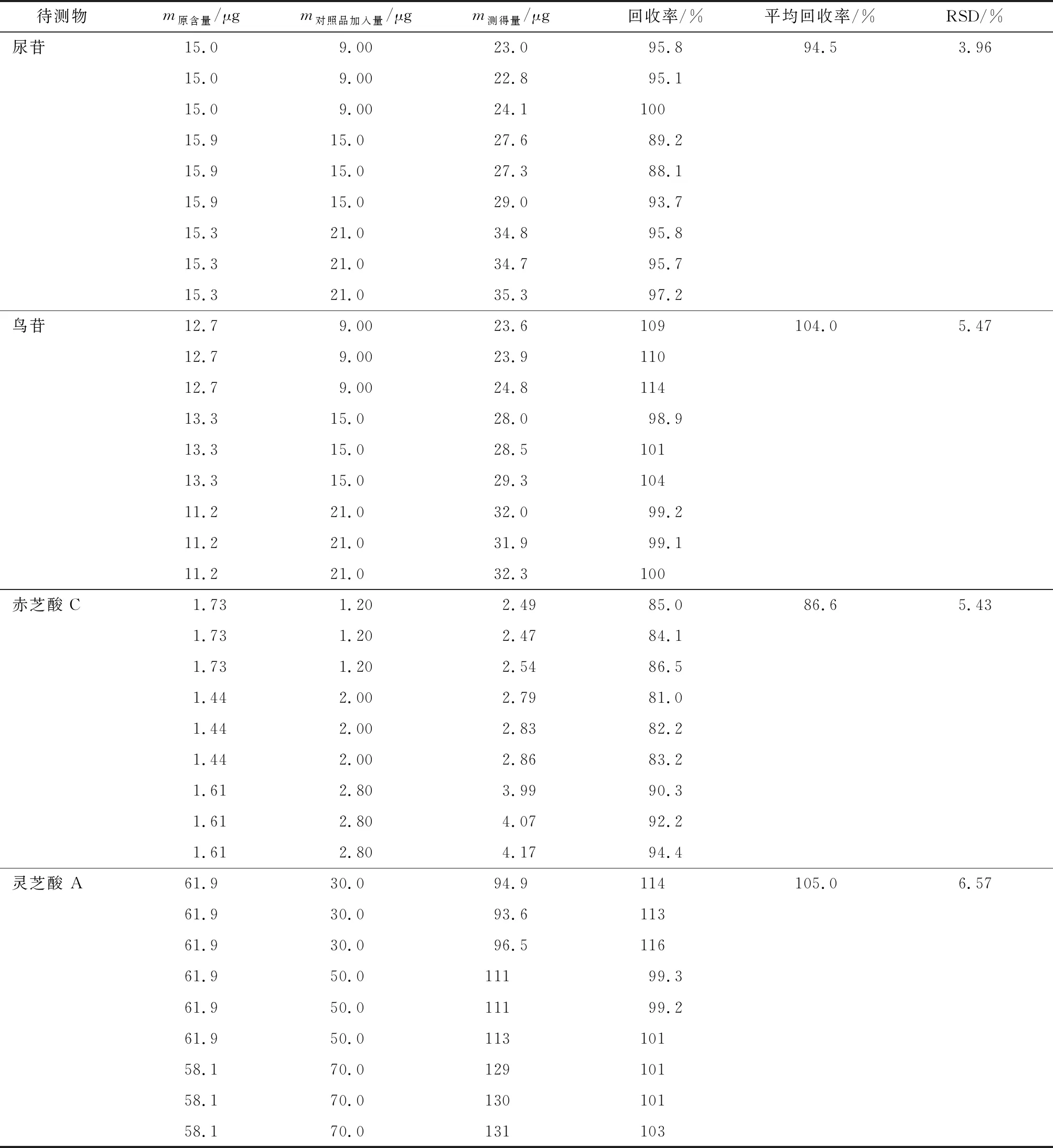
表3 加样回收率试验结果
2.7相对校正因子计算 在一定的线性范围内,待测成分的量(W)与检测器响应值(A)成正比,即W=f×A,选取待测成分中一组分k为参照物,建立组分k与其他组分m之间的相对校正因子,即fkm=fk/fm=(Wk×Am)/(Wm×Ak)。配置一定质量浓度的混合对照品溶液,精密吸取不同体积的同一混合对照品进样分析,分别计算不同进样体积下的fkm,然后计算平均值,并以此作为相对校正因子进行样品测定。按照1.4.9项下试验方法进样测定,以鸟苷为内参照物,分别计算鸟苷对尿苷、赤芝酸C和灵芝酸A的相对校正因子,相对校正因子的平均值分别为1.02,0.34,0.40,RSD值(n=3)均小于2%。
2.8一测多评法与外标法测定结果比较 采用外标法和一测多评法计算尿苷、鸟苷、赤芝酸C和灵芝酸A含量,结果见表4。
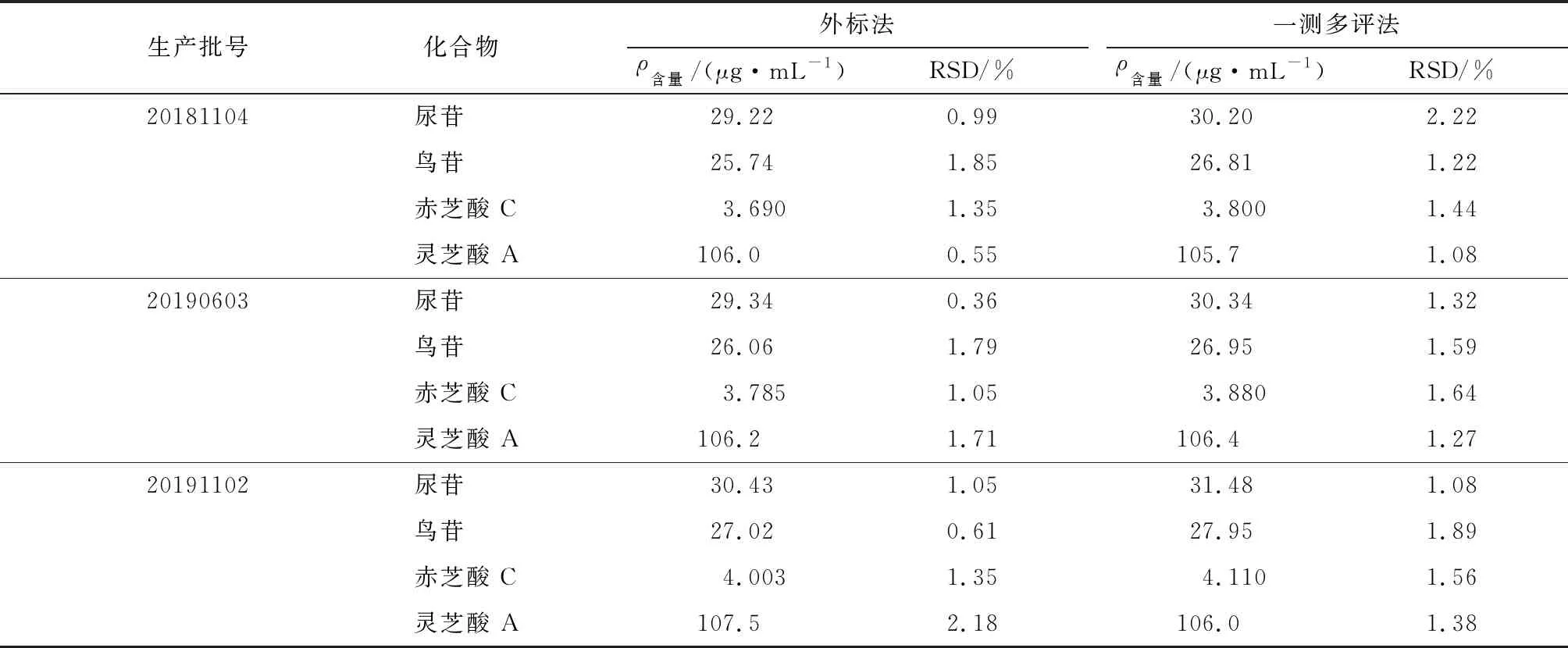
表4 外标法和一测多评法测定复方金线莲口服液中4种成分含量
将常规的外标法实测含量与一测多评法计算的含量采用向量夹角法进行比较,验证一测多评法用于多指标有效成分质量评价的准确性。夹角余弦值计算公式[16]如下:
本研究采用一测多评法和外标法测得4种有效成分的计算值和实测值之间的夹角余弦值均达到0.999 9,表明两种方法测得的含量没有显著性差异,说明一测多评法可用于复方金线莲口服液多成分质量评价的研究。
3 讨 论
本研究考察了不同有机相、pH及乙酸铵浓度对4种有效成分色谱分离的影响,结果表明流动相为甲醇-乙酸铵时,色谱峰出峰慢,目标峰与干扰峰不能得到较好的分离。在乙腈-乙酸铵(13 mmol/L)体系中加入乙酸(pH=4),各化合物色谱峰就可得到较好的分离。同时,本实验还对流动相流速、洗脱梯度进行了反复筛选优化,确定了流动相最佳梯度的洗脱程序。
核苷是生物细胞维持生命的重要物质,具有抗病毒、免疫调节、镇静中枢神经、调节心血管等多种药理生理活性[17],尤其是在抗乙型肝炎病毒方面具有明显的疗效[18],金线莲及灵芝中均富含核苷类物质尿苷和鸟苷[3,19]。三萜类化合物是灵芝的主要有效成分,具有抗肿瘤、抗病毒、保护肝脏等多种药理活性[20],灵芝酸A和赤芝酸C是其中具有代表性的成分。
采用一测多评校正因子对样品进行含量计算,在不同波长下偏差较大,因此选定适合的波长对降低测定误差很关键。经二极管阵列检测器分析,尿苷、鸟苷、赤芝酸C和灵芝酸A的最大吸收波长分别为261,251,251和259 nm,综合考虑4个成分,结果发现在254 nm处4个成分均有较大吸收且色谱峰分离度良好,达到色谱分析要求,因此选用254 nm作为检测波长。鸟苷性质稳定,不与其他成分反应,也不受口服液中其他成分干扰,容易购买,以此为参照物,各成分的相对校正因子稳定,重现性良好,且相比其他成分分离效果较好。本研究分别采用外标法和一测多评法对复方金线莲口服液中4种有效成分进行含量测定,结果使用向量夹角法进行比较分析,2种方法对同一样品的测定结果的夹角余弦值为0.999 9,表明2种测定方法所得结果具有较好的重现性、准确性,说明一测多评法可用于复方金线莲口服液中尿苷、鸟苷、赤芝酸C和灵芝酸A的含量测定。本研究的测定方法为复方金线莲口服液保肝功效的质量标准提供了参考依据,同时,也为复方金线莲口服液的药理活性研究和临床应用研究提供物质基础。
