Ca(II)-Gd(III)四核配合物的晶体结构与发光性质
2020-10-14叶发兵朱立红陈砚美
汪 洋,叶发兵,朱立红,陈砚美
(黄冈师范学院化学化工学院 催化材料制备与应用湖北省重点实验室,湖北 黄冈 438000)
异金属配合物在发光电子器件[1-2]、荧光探针[3-4]、磁性材料[5-6]、气体分离[7-8]和多相催化[9-10]等领域有着广泛的应用优势,而受到广泛的研究关注.稀土金属配合物具有独特的光学、电学和磁学性质,特别是作为稀土发光材料[11-15].近年来,含稀土金属离子的异金属发光配合物受到了化学家们的重视,其中,Zn-Ln的3d-4f异金属配合物已被大量报道,它们大部分具有优良的光学性能[16-18].Zn(II)的最外层电子构型为3s23p63d10,为闭壳层电子构型,碱土金属Ca(II)离子的最外层电子构型3s23p6,也具有闭合壳层电子结构,因此当碱土金属Ca(II)离子与稀土金属离子形成的异金属配合物,也应具有较好的发光性能.目前,已有一些具有良好发光性能的“碱土金属-稀土金属”(AE-Ln)异金属配合物被报道出来[19-23].可见,碱土-稀土异金属发光配合物具有较大的研究意义.
在之前的工作中,本课题组已经报道出一系列碱土-稀土异金属的配位聚合物,它们均呈现出了稀土离子的优良发光性能[24-26].本文报道了一个四核的异金属配合物[Gd2(pydc)6Ca2(H2O)10]·2Him·6H2O (1,H2pydc=吡啶-2,6-二甲酸; Him=质子化的咪唑),通过单晶衍射测定了其分子结构,并研究了它的红外光谱、差热分析光谱和荧光发射光谱性能.
1实验部分
1.1仪器试剂
化学试剂都是分析纯.表征用仪器为:SHIMADZU XRD-6100粉末衍射仪;Carol-Erba EA1110 CHNO-S元素分析仪;Nicolet NEXUS-6700傅立叶红外光谱仪(用KBr压片);Bruker SMART APEX ΙΙ单晶衍射(Mo Kα射线,λ=0.071073 nm);TA SQT-Q600热重分析仪;RF-5301荧光分光光度计.
1.2配合物的合成
依次称取吡啶-2,6-二甲酸(0.033 6 g,0.2 mmol)、Gd(NO3)3·6H2O (0.022 5 g,0.05 mmol)、CaCl2(0.010 8 g,0.1 mmol)和咪唑(0.047 8 g,0.7 mmol)放入一个耐高温的玻璃小瓶(8 mL)中,加入2 mL的乙醇/水(1∶2,V∶V)溶剂,密封.将混合物放在烘箱中,在自生压力下120 ℃加热72 h.冷却后,得到无色块状晶体.产量为:0.023 3 g (51.5%,按照Gd元素计算).元素分析理论值/%:C,31.82; H,3.34; N,7.73; 实验值/%:C,31.63; H,3.45; N,7.69. IR (cm-1,KBr):3 439(w),3 155(w),2 005(w),1 625(s),1 587(s),1 568(s),1 436(s),1 376(s),1 279(s),1 185(m),1 073(s),1 021(s),919(s),859(s),769(s),729(s),694(s),663(s),635(s),589(s),416(s).
1.3配合物1的结构表征
选取配合物1的单晶,用Bruker SMART APEX-II CCD衍射仪收集衍射数据,采用的ω-2θ扫描模式,测试温度为296(2) K,测试采用Mo Kα射线进行.晶体结构是通过SHELXS-97程序[27]计算,采用SHELXL-2016[28]的全矩阵最小二乘法F2进行结构精修.配合物1中的氢原子采取了两种加氢方式,吡啶环和质子化的咪唑上的氢原子通过理论计算加氢,水分子上的氢原子采用傅立叶电子密度分布图加氢.配合物1的晶体学参数见表1.配合物1的键长键角和氢键数据见表2~表4,其单晶结构数据(CCDC号:1965473)已保存至剑桥晶体数据库中心.

表1 配合物1的晶体学参数Tab. 1 Crystal data and structure refinement for complex 1

续表1

表2 配合物1的部分键长Tab.2 Selected bond lengths for 1

表3 配合物1的部分键角Tab.3 Selected bond angles for 1
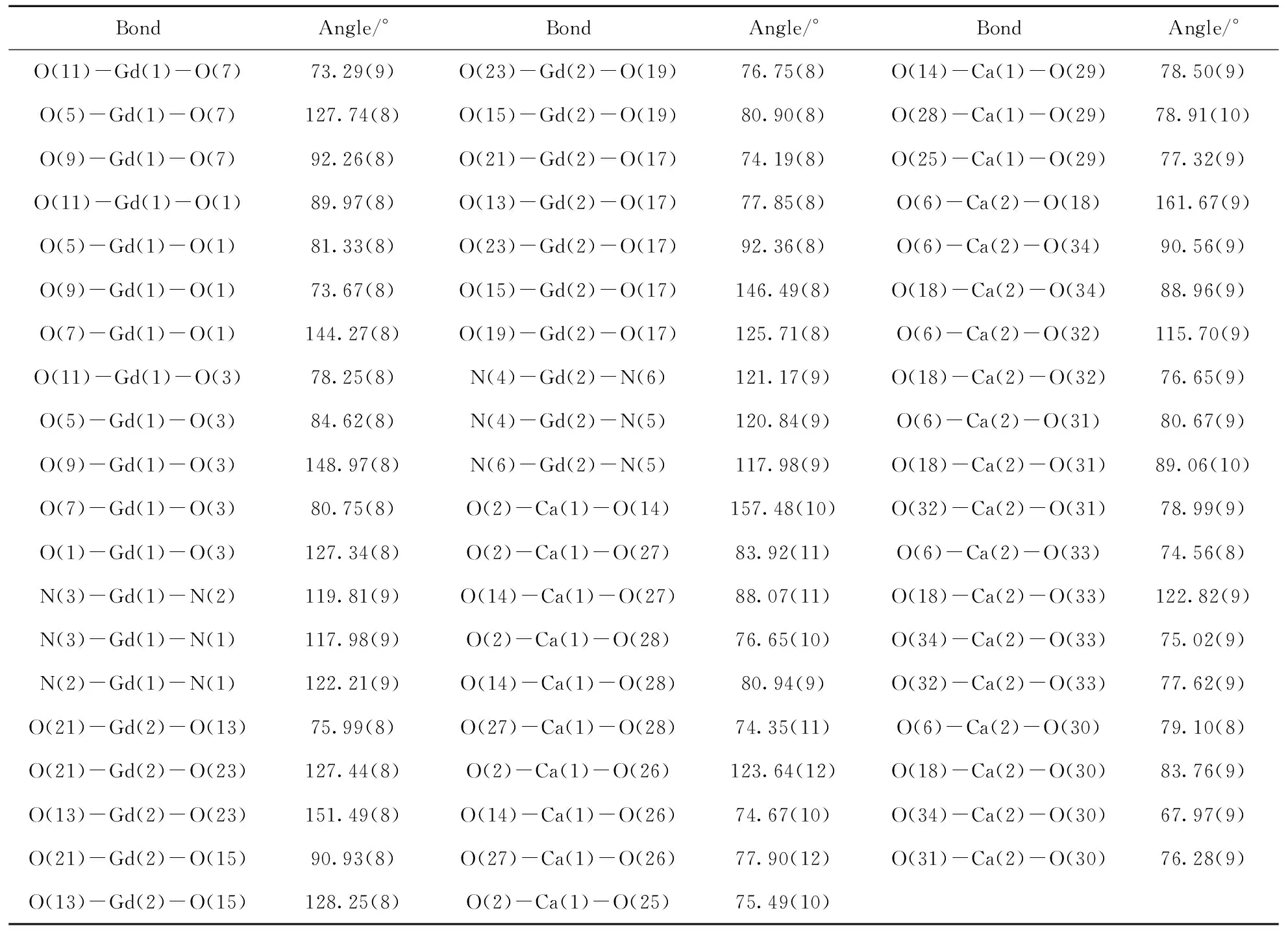
续表3

表4 配合物1的氢键的键长和键角Tab.4 Hydrogen bond lengths and bond angles for 1

续表4
2结果与讨论
在配合物的合成中,要控制Gd(NO3)3·6H2O与CaCl2的物质的量为1∶2,保持CaCl2过量一倍,两个金属离子易于同时发生配位,这可能是由于Ca(II)离子的配位竞争能力弱于Gd(III)离子的原因.此外,咪唑的加入对配合物的合成起到了关键的作用,咪唑在体系中起到了有机碱的作用,当咪唑与配体的物质量之比控制在7∶2时,能得到较纯的配合物1的单晶.当咪唑的量减少到0.5 mmol以下时,容易得到已被报道的单金属钙的配合物[Ca2(pydc)2(H2O)3]n(简写为Ca-pydc)[29];当咪唑的量增加时,体系容易得到较多的沉淀,不能得到纯净的配合物1的单晶.
2.1晶体结构
单晶衍射数据显示,配合物[Gd2(pydc)6Ca2(H2O)10]·2Him·6H2O (1)为Ca(II)、Gd(III)同时配位的异金属四核配合物,属于三斜晶系,P空间群,其中,[Gd2(pydc)6Ca2(H2O)10]2-为配合物离子,质子化的咪唑(Him+)为抗衡阳离子,另外还有六个游离的水分子.如图1所示,两个Gd(III)离子均为九配位,与三个吡啶-2,6-二甲酸根上的六个氧原子和三个氮原子配位,形成三帽三棱柱结构[Gd(pydc)3]3-(图2).Gd-O的键长范围为0.240 7(2)~0.252 2(2) nm,Gd-N的键长范围为0.251 2(3)~0.255 6(3) nm,与文献中报道的Gd-O和Gd-N键长相似[16,19].Ca1和Ca2离子均为七配位,Ca1分别与两个配体上的两个羧基O原子(O2和O14)和五个水分子(O25,O26,O27,O28,O29)配位,形成单帽三角棱柱体结构(图2).Ca2与Ca1的配位环境相似,也是与两个配体上的两个羧基O原子(O6和O18)和五个水分子(O30,O31,O32,O33,O34)配位,形成单帽三角棱柱体结构.其中,Ca-O的键长范围为0.2325(2)~0.2557(3) nm,与文献报道的Ca-O键键长一致[20,26,29].两个[Gd(pydc)3]3-结构通过四个配位的羧基(O1-C6-O2,O5-C13-O6,O13-C27-O14,O17-C34-O18)与Ca1和Ca2离子相连,形成一个环状的四核[Gd2(pydc)2Ca2(H2O)10]2-配合物阴离子结构.每个[Gd2(pydc)6Ca2(H2O)10]2-配合物阴离子之间通过O-H…O和C-H…O氢键相连,形成沿bc平面伸展的二维网状结构(图3).咪唑阳离子通过N-H…O和C-H…O氢键与相邻的二维网结构连接,形成三维超分子网状结构(图4).游离的水分子分布在三维超分子网状结构中,并通过O-H…O和C-H…O氢键与三维超分子网状结构连接.详细分子间氢键信息见表3.

图1 配合物1的50%热椭球图Fig.1 The ORTEP structure of complex 1 with 50% probability

图2 Ca1、Ca2和Gd1离子的配位多面体Fig.2 The coordination polyhedron of CA1,Ca2 and Gd1 cations
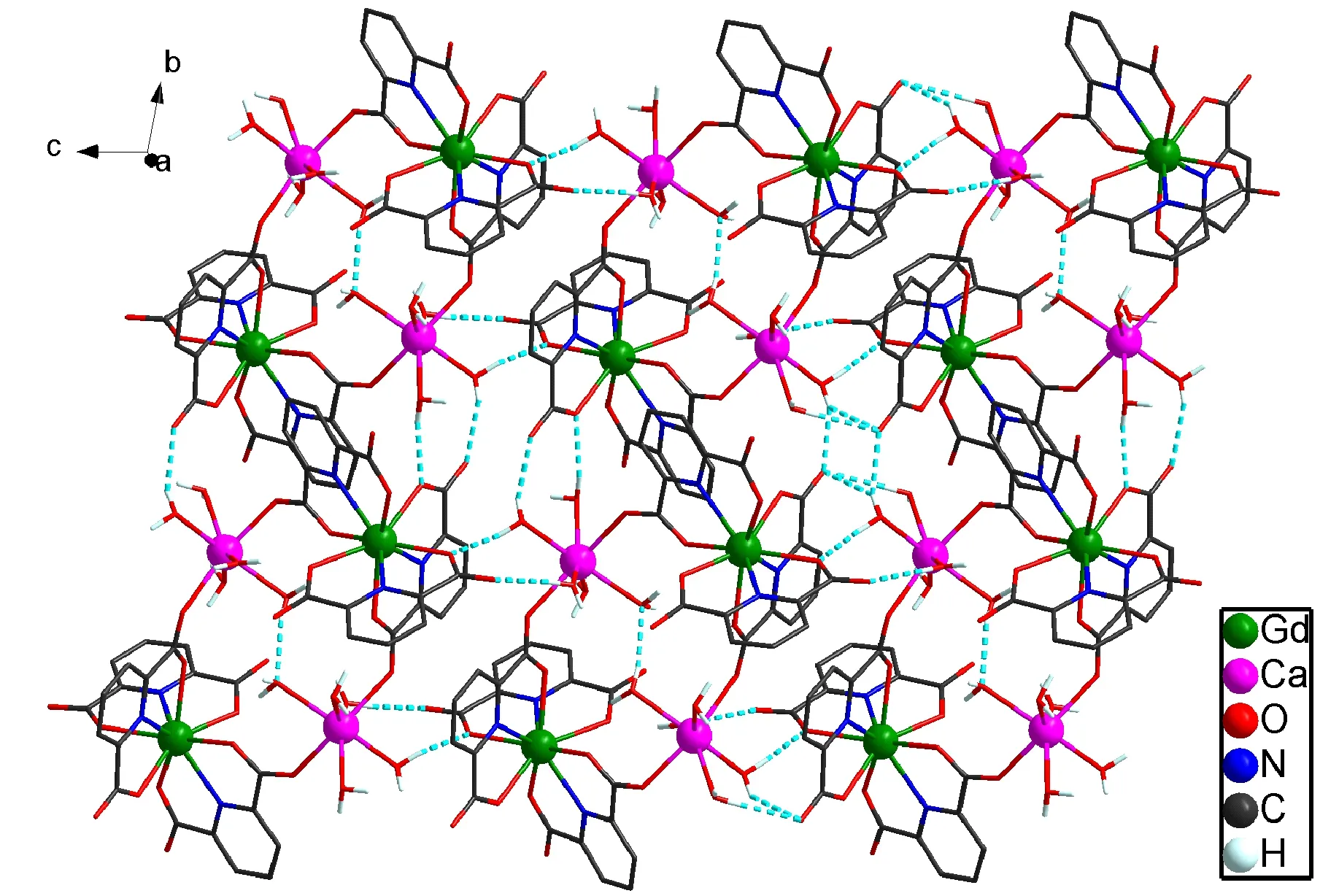
图3 bc平面的配合物1的二维氢键图Fig.3 2D Hydrogen bonds interactions of 1,in bc plane

图4 沿b轴看配合物1的三维氢键图Fig.4 3D Hydrogen bonds interactions of 1,viewed along b-axis
2.2热稳定性分析
在N2气氛下对配合物1的热稳定性进行了研究.从图5中可以看出,配合物1的失重是连续的,从室温到200 ℃之间,对应的重量损失为5.9%(理论值为5.94%),对应于六个游离水分子的失去.超过200 ℃之后,配合物有较大的失重,并伴随大量吸热,说明配合物逐渐分解.配合物的失重过程对应于热流曲线,其在200 ℃之前有一个小的吸热峰,说明失去六个游离水分子时为吸热反应,接着从200 ℃到340 ℃之间有一个较大的吸热峰,说明配合物的分解过程为吸热过程.高于340 ℃之后,又有一个平缓的放热峰,说明340 ℃之后,配合物的分解过程为放热过程.配合物1从室温开始失重,说明配合物1中游离水分子的稳定性不太好,但配合物的分解温度高于200 ℃,可见配合物的骨架结构相对稳定.

图5 配合物1的差热分析图谱Fig 5 TGA-DSC diagram for compound 1
2.3红外光谱
以溴化钾为载体,在4 000~400 nm范围内测定了配合物1的红外光谱.红外光谱数据显示3 439 cm-1和3 155 cm-1周围的宽频带,可以归因于γ(O-H)和γ(C-H)的伸缩振动,揭示了水分子和吡啶环的存在.羧基的伸缩震动在波数1 625 cm-1和1 587 cm-1处.波数1 436和1 376 cm-1处的尖锐峰,归属为吡啶环骨架的伸缩振动.1 279 和1 185 cm-1处为C-O键的面内弯曲振动,1 073,1 021和859 cm-1归属为吡啶环上C-H键的面外弯曲振动.729 和694 cm-1归属为Ca-O键的伸缩振动,663和 589 cm-1归属为Gd-O键的伸缩振动,通过红外峰波数可以看出配体与Gd(III)和Ca(II)均发生了配位[19-20,26].
2.4荧光性质
本文测定了配合物[Gd2(pydc)6Ca2(H2O)10]·2Him·6H2O (1)的固态荧光性质,并于配体吡啶-2,6-二甲酸和已报道的配合物[Ca2(pydc)2(H2O)3]n(简写为Ca-pydc)[29]的荧光进行了对比.以激发波长为327 nm对配合物1进行激发时,可以看到配合物1在470 nm处出现最强发射峰,呈现出亮蓝色荧光(图6).而配体吡啶-2,6二甲酸和配合物[Ca2(pydc)2(H2O)3]n的最大激发波长分别为363 nm和310 nm,以它们的最大激发波长测的它们分别在405 nm和421 nm处出现最强发射峰.通过对比可以看出,配合物1和配合物[Ca2(pydc)2(H2O)3]n的最大荧光发射波长相对于配体都发生了红移,这是金属离子的配位导致的配合物的荧光发射峰红移.金属离子Ca(II)和Gd(III)都比较稳定,难以得失电子,因此配合物的荧光归属为金属离子干扰下的配体到配体的电荷转移(LLCT)跃迁[30-32].配合物1的最大荧光发射峰对应的波长比配合物[Ca2(pydc)2(H2O)3]n的最大发射波长略有增加,可以看出Ca(II)和Gd(III)离子的同时配位,与单金属Ca(II)配位相比,对荧光发射波长的影响不同,这不仅与配位的金属离子的种类有关系,还有金属离子的配位方式有关.配合物1显示出明亮的蓝色发光,可作为蓝光材料.

图6 H2pydc (λex=363 nm)、Ca-pydc (λex=310 nm)和配合物1(λex=327 nm)的荧光发射光谱图Fig.6 The emission spectrum for H2pydc ligand,Ca-pydc and complex 1
3小结
本文合成了一个Ca(II)-Gd(III)异金属四核配合物[Gd2(pydc)6Ca2(H2O)10]·2Him·6H2O (1),并通过X-射线单晶衍射技术确定了配合物的主体结构为四核配合物阴离子,每个配合物阴离子之间通过O-H…O和C-H…O分子间氢键相连形成二维平面结构,每个平面之间通过和咪唑阳离子链接形成三维超分子结构.游离的水分子分布在三维超分子网状结构中,并通过O-H…O和C-H…O氢键与三维超分子网状结构连接.热稳定性结果表明,配合物1中的游离水分子很容易失去,其失重过程伴随吸热,超过200 ℃之后配合物开始分解,分解过程伴随较大的吸热.从红外光谱数据上可以看到配体与金属Ca(II)和Gd(III)离子的都发生了配位.室温固态荧光发射光谱显示,配合物1的最大发射峰对应的波长与配体相比发生了红移,它呈现出明亮的蓝色荧光,可作为潜在的蓝光材料.
