悬浮固化液相微萃取高效液相色谱法测定环境水样中磺胺类药物
2020-09-26沈杨银饶桂维蔡婕妤卜兴明傅煜豪
沈杨银,饶桂维,蔡婕妤,卜兴明,傅煜豪
(浙江树人大学生物与环境工程学院,杭州 310015)
磺胺类抗生素(Sulfonamides,SAs)属于人工合成的抗菌药物,基本化学结构为对氨基苯磺酰胺。由于具有抗菌谱广、化学性质稳定、价格便宜、使用方便等优点,SAs 被广泛使用于牲畜养殖业中[1]。根据统计,SAs 在世界各国的抗生素销售及使用量中均居于首位[2]。大量施用的SAs 会通过农田土壤地表径流冲刷渗滤、畜禽粪便排放、污水处理厂出水等途径最终进入水体环境[3]。SAs 在动物体内的代谢时间较长,容易残留在动物产品当中,水体环境中的SAs 会通过食物链进入人体,在人体内蓄积,造成泌尿、免疫和造血系统紊乱,诱发皮炎等过敏反应并具有致癌性,还会导致人体出现急性或慢性中毒,从而威胁人体健康,因此研究磺胺类药物的检测方法很有重要。
目前,检测磺胺类药物残留的方法主要有气相色谱–质谱(GC–MS)法[4]、高效液相色谱(HPLC)法[5]、液相色谱–串联质谱(LC–MS)法[6]、毛细管电泳法[7]、酶联免疫分析法[8]等。样品前处理是分析过程的重要组成部分。传统的样品前处理技术有固相萃取(SPE)[9]、固相微萃取(SPME)[10]、液液萃取(LLE)[11]、液相微萃取(LPME)[12]。这些前处理技术普遍存在以下缺点:处理时间长,试剂用量大,萃取速度慢,仪器价格高,操作繁琐,高毒性,实现自动化困难,难以满足对痕量污染物监测的要求[13]。LPME 技术逐渐发展,分为单液滴微萃取(SDME)[14]、中空纤维液相微萃取(HFLM)[15]、分散液–液微萃取(DLLME)[16]、悬浮固化液相微萃取(SFO–LPME)。
单液滴微萃取技术是将有机萃取剂悬挂于针头上进行萃取的技术,人工难以控制,难以达到要求的精确度与准确度,此技术的萃取效率由液滴的量来决定,但较大液滴在针头上非常容易掉落,影响萃取效率。中空纤维液相微萃取技术中纤维孔隙很小,孔径一般为0.2 μm,容易存在样品堵塞微孔的现象,该技术操作较为繁琐,萃取速率慢,难以实现自动化。分散液–液微萃取技术样品溶液若含有颗粒和大分子会影响萃取效果,该技术常选用卤代烃作为有机萃取剂,而卤代烃毒性较强,容易对实验人员造成伤害,且对环境有严重污染。
悬浮固化液相微萃取技术由Yamani 等[17]于2007 年提出,具有萃取时间短、成本低、操作简便、有机溶剂用量少、对环境友好等优点,是一种新型的样品前处理技术,在与HPLC 仪、原子吸收光谱(AAS)仪、GC 仪、高效毛细电泳(HPCE)仪等联用后,可以对目标物进行定性和定量分析,在痕量分析领域拥有广阔的应用前景。悬浮固化液相微萃取通常使用磁子搅拌进行萃取,萃取过程要求实验人员具有一定的专业技能且耗时较长。笔者采取悬浮固化液相微萃取技术与液相色谱联用,采用涡旋辅助微萃取,建立了以悬浮固化液相微萃取技术测定环境水样中磺胺类药物的新方法。
1 实验部分
1.1 主要仪器与试剂
高效液相色谱仪:P230II 型,配UV230II 型紫外可见检测器、LU230II 型低压梯度混合器、P230II 型高压恒流泵,大连伊利特分析仪器有限公司;
超纯水器:UPWS–1–60D 型,杭州永洁达净化科技有限公司;
涡旋混合器:SW–80A 型,宁波新芝生物科技股份有限公司;
台式高速冷冻离心机:GL–20G–H 型,上海安亭科学仪器厂;
电子天平:MS205DU 型,瑞士梅特勒–托利多集团;
数控超声波清洗器:KQ5200DE 型,昆山市超声仪器有限公司;
隔膜真空泵:GM–0.33 型,天津市津腾实验设备有限公司;
磺胺嘧啶(SD)、磺胺二甲嘧啶(SM2)、磺胺对甲氧嘧啶(SMD)标准样品:纯度均不小于98%,西格玛奥德里奇(上海)贸易有限公司;
乙腈、甲醇:色谱纯,美国天地试剂公司;
正十二醇:纯度不小于99%,上海麦克林生化科技有限公司;
磷酸:分析纯,国药集团化学试剂有限公司。
1.2 标准溶液的配制
分别准确称取60 mg SD 标准样品,100 mg SM2 标准样品,140 mg SMD 标准样品,用乙腈溶解并定容至100 mL 容量瓶中,配制成SD 质量浓度为600 mg/L,SM2 质量浓度为1 000 mg/L,SMD质量浓度为1 400 mg/L 的标准储备溶液,置于4℃冰箱中备用。使用前用乙腈稀释成SD 质量浓度为3.75~60 mg/L,SM2 质量浓度为6.25~100 mg/L,SMD 质量浓度为8.75~140 mg/L 的系列混合标准工作溶液。
1.3 模拟水样的配制
用烧杯称取适量SAs(SD,SM2,SMD),以乙腈溶解,转移至1 L 容量瓶中,用去离子水定容,摇匀后配制成一定浓度的模拟水样。
1.4 样品采集与前处理
将采集的水样储存于50 mL 离心管中,在实验室冰箱中冷藏保存过夜,次日,取摇晃均匀后的10 mL 水样,加入0.8 mL 正十二醇,涡旋1 min,在4 000 r/min 转速下离心5 min,将离心后的溶液冰水浴5 min,用药匙将固化后的正十二醇取出,置于干净的离心管中,待正十二醇融化后加入0.8 mL 甲醇,将样品溶液过0.22 μm 滤膜,进样至高效液相色谱仪中进行分析。
1.5 色谱条件
色谱柱:SinoChrom ODS–BP 型柱(4.6 mm× 200 mm,5 μm,大连伊利特分析仪器有限公司);柱温:30℃;流动相:体积分数为0.1%的磷酸水溶液–乙腈(90∶10),流量为1 mL/min;进样体积:10 μL;检测波长:270 nm。
2 结果与讨论
2.1 萃取剂的选择
悬浮固化液相微萃取要求萃取剂具有水溶性低、不易挥发、密度小于水、熔点接近或低于室温(10~30℃)、萃取容量高和对分析物测定没有干扰等特点。实验室经常使用正十一醇、正十二醇、正十六烷、溴代十六烷作为萃取剂。选择正十一醇和正十二醇进行试验,发现相同条件下正十一醇在萃取SAs 时冰浴后没有固化现象,有机相不能与水相分离,不能满足萃取要求,而用正十二醇萃取SAs时效果良好,故选择正十二醇作为萃取剂。
2.2 辅助乳化条件的选择
悬浮固化液相微萃取技术会利用涡旋和超声等形式辅助乳化,涡旋可以加快萃取剂与模拟水样之间的传质速率,加速乳化形成,缩短萃取时间。悬浮固化液相微萃取技术利用超声辅助时,萃取剂在超声辅助下以微小液滴的形式分散于水相中,与水相之间形成巨大的接触面,从而加快物质的转移,实现萃取目的。对超声辅助和涡旋辅助进行对比试验,结果如图1。由图1 可知,在相同条件下,涡旋比超声具有更高的提取率;并且在萃取过程中,涡旋比超声耗时更短,耗能更少;而且涡旋设备便于携带。故选择涡旋辅助乳化进行微萃取操作。
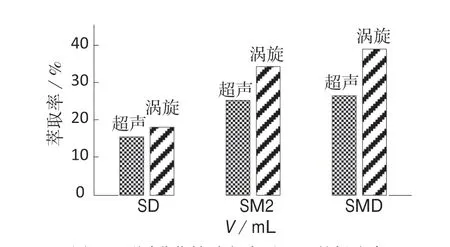
图1 不同乳化辅助方式下SAs 的提取率
2.3 萃取剂用量的优化
萃取剂用量是影响水样中SAs 提取率的重要因素之一,因为正十二醇与水相的接触面积与SAs向正十二醇的传质效率有很大关系,随着萃取剂用量的增加,SAs 的提取率也会随之提高。试验对比了0.3,0.4,0.5,0.6,0.8,1.0,1.1 mL 正 十 二 醇 萃 取10 mL 水样的提取率(如图2 所示)。试验结果表明,萃取10 mL 环境水样中的SAs,使用0.8 mL 正十二醇时提取率最高。由图2 可知,随着正十二醇用量的增大,SAs 的提取率逐渐升高,而正十二醇的最佳萃取剂用量为0.8 mL。当萃取剂用量超过0.8 mL时,随着正十二醇用量的增大,SAs 的提取率下降,这是过多的萃取剂起到稀释作用导致,故选择萃取剂最佳用量为0.8 mL。
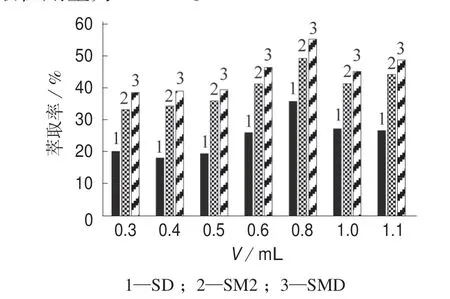
图2 不同萃取剂用量时的SAs 提取率
2.4 溶液pH 值对提取率的影响
溶液pH 对SAs 的提取率也有影响。调节水样pH,改变SAs 的状态,使其处于分子或离子状态,从而影响SAs 的提取率。分别使用硫酸溶液和氢氧化钠溶液调节溶液的pH,用pH 计测得模拟水样pH 为5.3。SD 易溶于稀盐酸、氢氧化钠溶液(SM2易溶于稀酸和稀碱溶液,SMD 易溶于稀酸)。pH为3.3~7.3时SAs的提取率如图3所示。由图3可知,溶液pH 为7.3 时,SM2 的提取率明显高于其它两种药物,溶液pH 为3.3 时,SMD 的提取率明显高于另外两种药物。这是因为SM2 易溶于稀碱,SMD易溶于稀酸,酸碱性试剂的加入有利于SM2,SMD在模拟水样中的溶解,从而提高提取率。试验结果表明,不调节模拟水样pH 时3 种SAs 的综合提取率最好,故选取pH 5.3 进行后续试验。
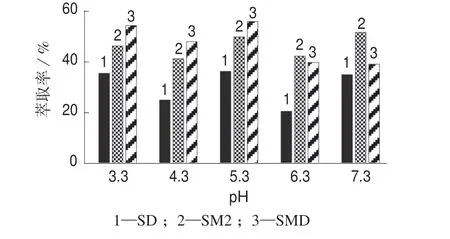
图3 不同pH 时的SAs 提取率
2.5 涡旋时间对提取率的影响
萃取时间是影响提取率的一项要素,使用涡旋的方法进行萃取试验,在涡旋过程中,足够的萃取时间可以使目标分析物最大程度地转移进入萃取剂中,提高SAs 的提取率。图4 展示了不同涡旋时间对应的SAs 提取率,由图4 可见,涡旋时间为15~60 s 时,SAs 提取率随着涡旋时间的增长而增大,在涡旋时间为60 s 时SAs 提取率达到最大,然后呈下降趋势。原因是涡旋时间太短会导致萃取剂与水样中的SAs 不能达到分配平衡,SAs 未能最大程度地进入正十二醇;涡旋时间过长则会造成乳浊液不稳定,SAs 提取率也会降低且浪费能量。综合考虑,选取最佳涡旋时间为60 s。
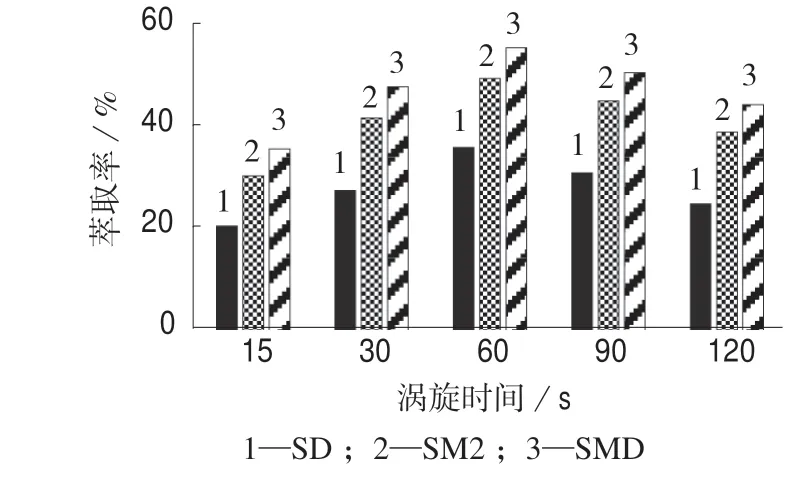
图4 不同涡旋时间时的SAs 提取率
2.6 离心时间对提取率的影响
分别选择离心时间为1,3,5,7,9 min,考察SAs的萃取率,试验结果如图5 所示。试验发现随着离心时间增加,SAs 的提取率起初随之提高,在离心时间为5 min 时达到最大,然后呈下降趋势。最初提取率随着离心时间增加而提高,可能是因为过短的离心时间导致萃取剂萃取完SAs 后与水样分层不够完全,冰浴后固化的十二醇结构松散,收集较为困难,会有所损耗,导致SAs 提取率较低。而随着离心时间的增加,SAs 提取率达到峰值后即形成动态平衡,而过长的离心时间会导致耗能增加。综合考虑,选取离心时间为5 min。
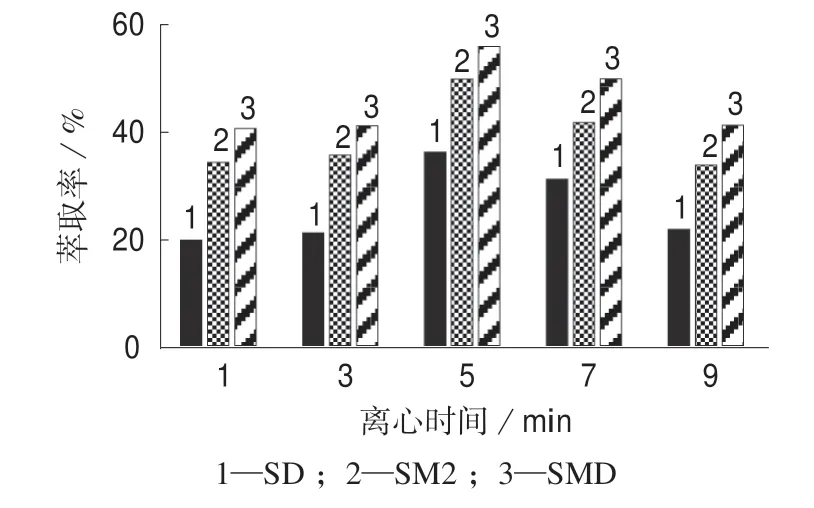
图5 不同离心时间时的SAs 提取率
2.7 离心转速对提取率的影响
在2 000~6 000 r/min 区间分别设置不同的离心转速,考察离心转速对SAs 提取率的影响,试验结果如图6 所示。试验结果表明,SAs 的提取率随着离心转速的上升而增大,在离心转速为4 000 r/min 时SAs 的萃取率达到极值,然后开始降低。这可能是由于离心转速过高导致SAs 分解,破坏了正十二醇的结构,从而使SAs 的提取率降低。为了使萃取SAs 的正十二醇与样品溶液充分分离,选则最佳离心转速为4 000 r/min。
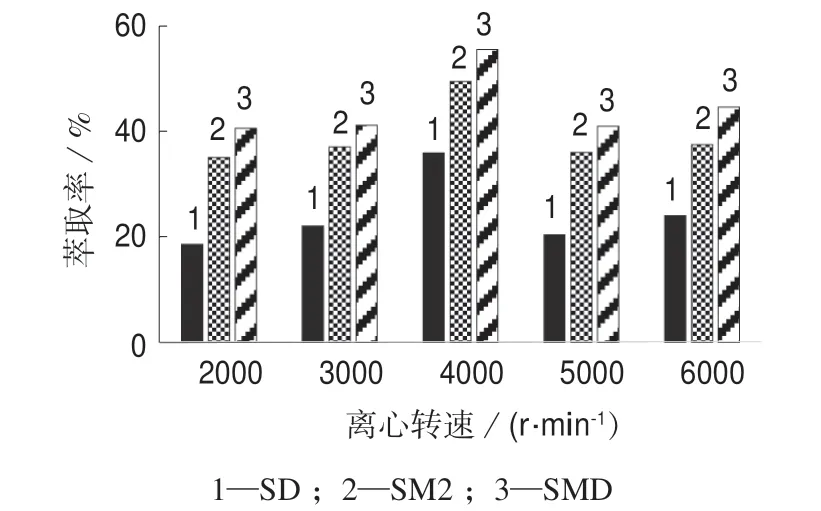
图6 不同离心转速时的SAs 提取率
2.8 线性范围、检出限与定量限
配制质量浓度分别为3.75,7.5,15,30,60 mg/L 的系列SD 标准工作溶液,质量浓度分别为6.25,12.5,25,50,100 mg/L 的系列SM2 标准工作溶液,质量浓度分别为8.75,17.5,35,70,140 mg/L 的系列SMD 标准工作溶液,在1.5 色谱条件下测定,以SAs的质量浓度(x,mg/L)为自变量、色谱峰面积(y)为因变量进行线性回归,计算线性方程、相关系数。
以信噪比(S/N)为3 时对应的标准溶液质量浓度为方法检出限,信噪比为10 对应的标准溶液质量浓度为定量限。
线性范围、线性方程、检出限和定量限列于表1。由表1 可知,SD,SM2,SMD 的质量浓度分别在3.75~60,6.25~100,8.75~140 mg/L 范围内线性关系良好,相关系数(r)均不小于0.999 2,SD,SM2,SMD 的检出限分别为0.48,0.72,0.92 mg/L,定量限分别为1.59,2.40,3.05 mg/L。

表1 3 种SAs 标准曲线的线性方程、相关系数、检出限及定量限
2.9 加标回收试验
分别取质量浓度为15,30,60 mg/L 的SD 溶液,质量浓度为25,50,100 mg/L 的SM2 溶液和质量浓度为35,70,140 mg/L 的SMD 溶液,按1.4 方法处理,在1.5 色谱条件下进行加标回收试验,平行试验5次,计算平均回收率和相对标准偏差,试验结果列于表2。加标样品色谱图如图7。由表2 可知,该方法SD,SM2,SMD 的精密度良好,SM2,SMD 回收率良好。SD 鉴于其本身几乎不溶于水的物理性质,使用该方法情况下损耗较大,导致其回收率较差。
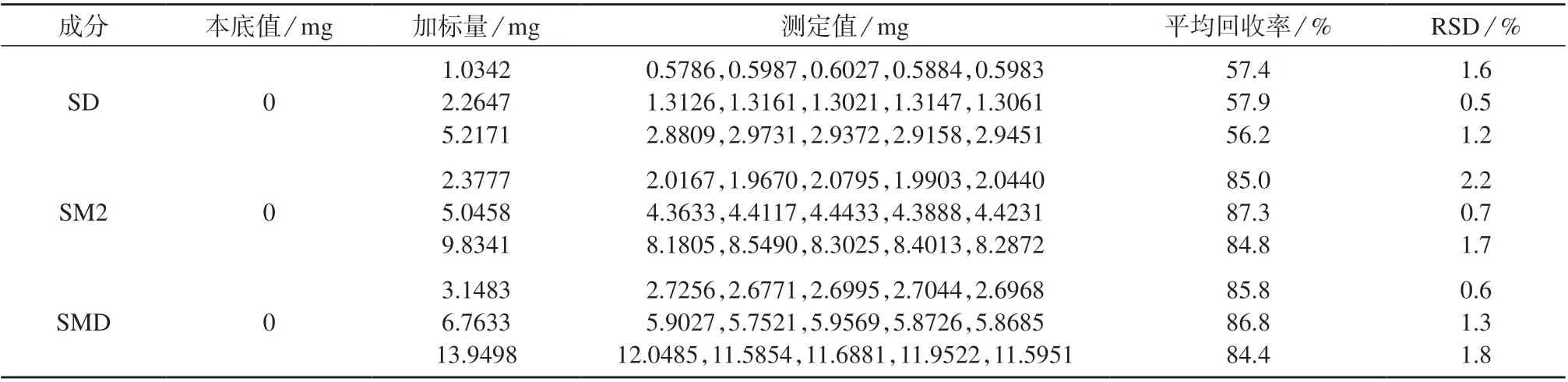
表2 加标回收试验结果
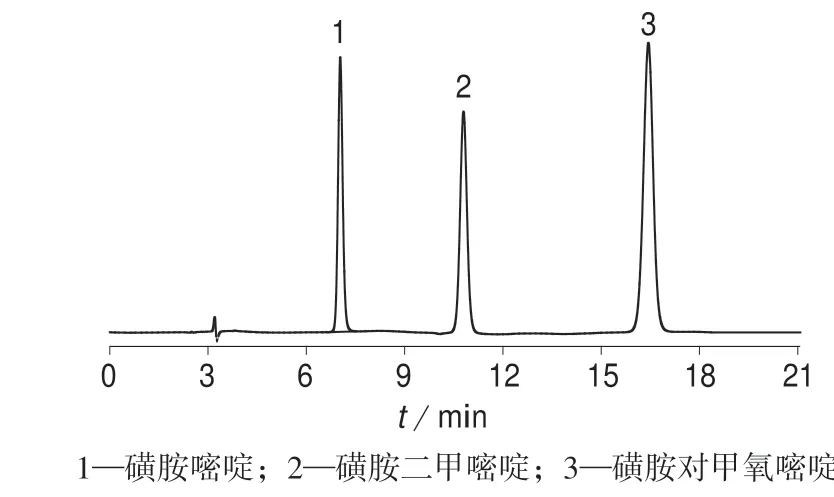
图7 加标样品色谱图
2.10 稳定性试验
吸取同一标准混合溶液10 μL,分别在0,2,4,6,8,10 h 于1.5 色谱条件下进样测定,记录色谱峰面积,计算峰面积测定值的相对标准偏差,SD,SM2,SMD 的色谱峰面积测定值的相对标准偏差分别为2.8%,1.8%,2.3%,表明该方法具有良好的稳定性。
3 结语
建立了一种悬浮固化液相微萃取–高效液相色谱联用测定环境水样中磺胺类药物的方法,实验过程中以正十二醇作为萃取剂采取涡旋辅助微萃取,该方法试剂用量少,操作简便,成本低廉,对环境友好,准确性、重现性好,可以满足样品分析的要求,为测定磺胺类药物在环境水样中的含量提供新思路和新的技术支持。
