硫代核苷类似物的合成研究进展
2020-09-21崔琳琳邢志华刘颖杰申光焕
李 洋, 崔琳琳, 邢志华, 刘颖杰, 袁 甜, 申光焕*
(1. 哈尔滨商业大学 a.药学院; b.黑龙江省预防与治疗老年性疾病药物研究重点实验室,黑龙江 哈尔滨 150076)
在过去对核苷类似物的研究中,发现此类化合物中的大部分具有抗肿瘤、抗病毒等[1-3]生理活性。目前已经上市的和处于临床试验的抗病毒药物绝大多数属于核苷类似物,其作为抗病毒药物在抗病毒的化学治疗中扮演着重要的角色[4-5]。核苷类似物可作为天然脱氧核苷酸的类似物嵌入病毒DNA双链,由于其缺乏3′-OH而导致逆转录反应终止[6-7]。然而,核苷类药物的耐药性,毒副作用大,活性不理想等[8]因素阻碍着核苷类药物的发展。对于天然核苷的结构改造、结构修饰以增加药物活性、降低药物毒副作用是寻找新药的重要手段。从首次发现碳环核苷化合物至今,已经有一些五元碳环核苷从天然产物中分离,并表现出显著的生物学活性[9]。为了筛选稳定有效的先导化合物,许多的核苷类似物被相继合成,并相继开发成药物[10],核苷类似物的合成以及生物活性的筛选已经成为目前核苷新药研究领域新的的热点。
Scheme 1
核苷通常定义为DNA或RNA亚基,并且由碱基部分如腺嘌呤,胸腺嘧啶,鸟嘌呤,胞嘧啶和尿嘧啶以及糖部分如D-核糖或D-脱氧核糖组成。糖修饰的核苷是抗癌和抗病毒药物开发的主要合成靶标[11]。其中呋喃糖环中的氧通过生物电子等排原理[12]被硫原子取代的4′-硫代核苷因其生物学特性,具有如抗病毒[13-14]和抗肿瘤[15]活性而受到特别关注。硫和氧属于元素周期表中的同一族,它们的大小和电负性不同。X-射线晶体学分析表明4′-硫代核苷采用与相应的4′-氧代核苷具有相同的构象,表明4′-硫代核苷与4′-氧代核苷一样可以是具有良好活性的临床药物。已知许多实例,在糖环中引入硫原子代替氧导致活性增加,如恩曲他滨和拉米夫定是用于治疗HIV的两种硫代核苷酸抗病毒药,6-硫鸟嘌呤和其他天然核碱基的硫代类似物对增殖细胞具有高度亲和力,其中一些已被用于治疗癌症,白血病和心绞痛。对于本次新型冠状病毒, 瑞德西伟也展现出良好的活性,表明三磷酸鸟苷衍生物可用作抗新型冠状病毒肺炎的特异性抑制剂[21-23]。此外,4′-硫代核苷具有更稳定的糖基键和针对各种病毒或细胞酶的代谢稳定性的提升等固有的优点。尽管上面提到了4′-硫代核苷的优点,但合成关键中间体4-硫代核糖的困难[24],在过去的研究中,报道了许多的4′-硫代核苷类似物的制备合成,使得能够对各种4′-硫代核苷进行构效关系的研究。
本综述根据4-硫代核糖(2′位和3′位的羟基取代情况)的不同取代,大致分为三类:具有糖构型的4′-硫代核苷、2′或3′-脱氧-4′-硫代核苷和2′,3′-双脱氧-4′-硫代核苷,对近年来各类4′-硫代核苷类似物的合成研究进展进行了简要回顾。
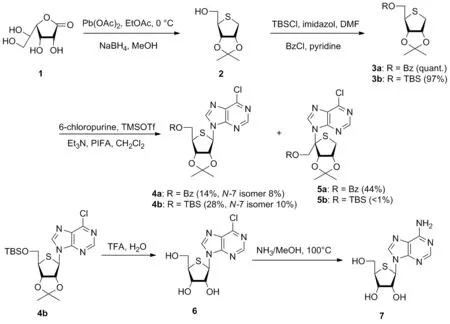
2008年,Nishizono等[25]通过合成硫代核糖进而合成出糖基含硫的核苷类似物,通过对Jeong等[26]的方法改进,用D-葡萄糖-γ-内酯1为底物,合成出1,4-脱水-2,3-异亚丙基-4-硫代D-核糖醇2,接着对2的羟基进行保护得到4和5,去保护基团后得到4′-硫代腺苷7,产率为59%(Scheme 1)。
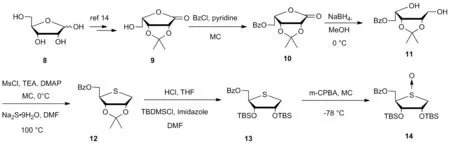
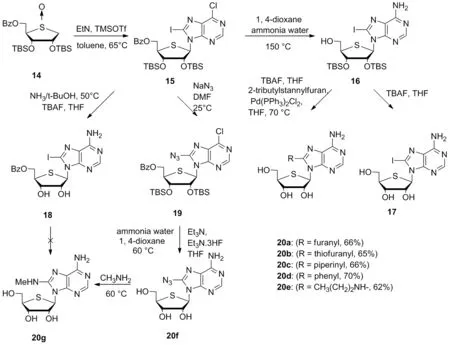
2016年,Qu等[27]设计合成出一系列作为具有潜力的HSP 90抑制剂的C8-取代-4′-硫代核苷类似物,并且对其抗肿瘤的活性予以研究。采用Pummerer反应或Vorbrüggen缩合反应和核碱基C8位的官能化通过Stille偶联或亲核芳香取代反应作为关键步骤,从D-核糖成功合成了一系列C8-取代-4′-硫代腺苷类似物20a~20g,17和18。对于C8取代的4′硫代腺苷衍生物20a~20g的合成,首先从D-核糖合成糖基供体(14, Scheme 2)。D-核糖8转化为L-莱索内酯衍生物9[28],再得到4-硫糖衍生物12[29]。使用Pummerer缩合反应和C8改性作为关键步骤完成C8-取代-4′-硫代腺苷(20a~20g, Scheme 3)。测定化合物的HSP 90抑制活性,发现它们在100 μM时无活性。然而,8-碘衍生物17和18表现出有效的抗癌活性,表明不同的作用机制与其生物活性相关。
Scheme 2
Scheme 3
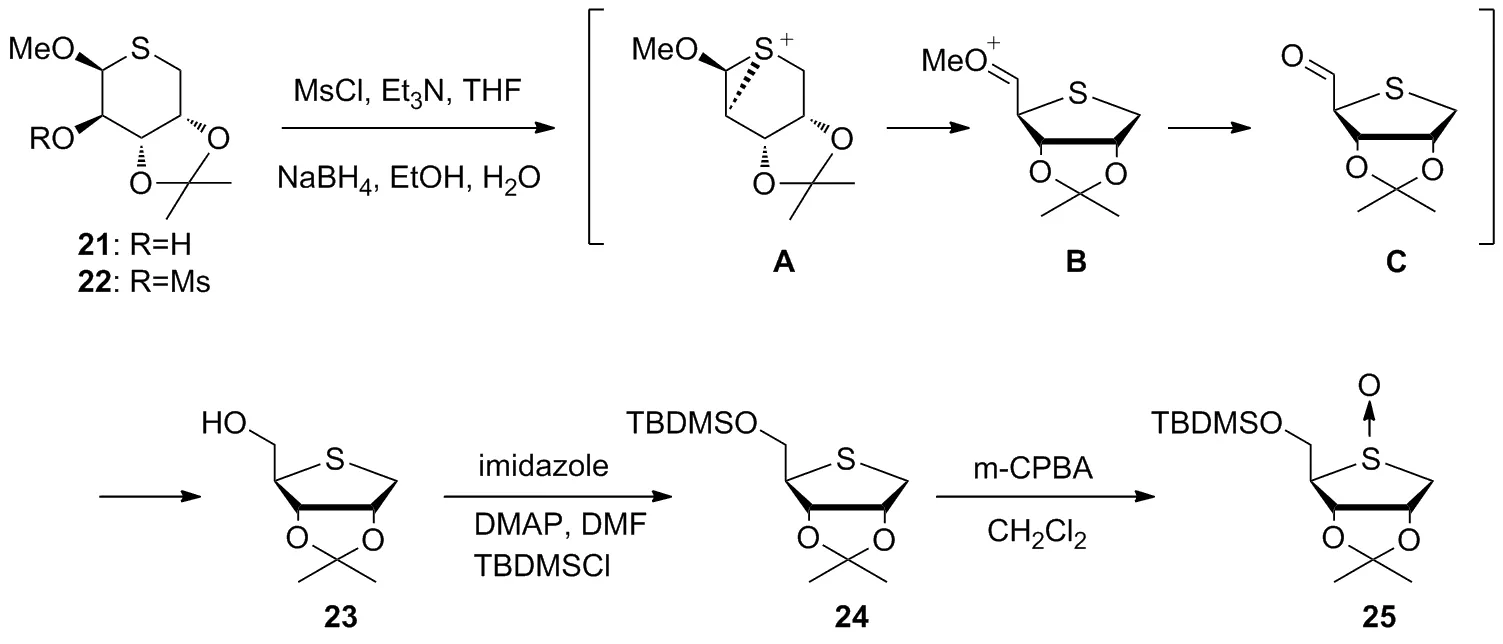
2017年,Wakamatsu等[30]设计并详细描述了4′-硫代核苷的的合成路线。1,4-脱水-2,3-O-异亚丙基-4-硫代核糖醇是合成4′-硫代核苷的关键中间体,通过多个步骤从L-阿拉伯糖中获得,关键中间体氧化后,将亚砜在存在全硅烷基化的核碱基的条件下进行Pummerer型硫糖基化反应,从而以高收率和β-选择性获得4-硫代核糖核苷。在还原条件下将甲磺酸盐22直接转化为所需的4-硫代核糖。反应平稳进行以提供高收率的23,并且通过5-O-甲硅烷基化和随后的m-CPBA氧化将23转化为5-O-甲硅烷基化的亚砜(25, Scheme 4)。
Scheme 4
Scheme 5
Scheme 6

Scheme 7
Scheme 8
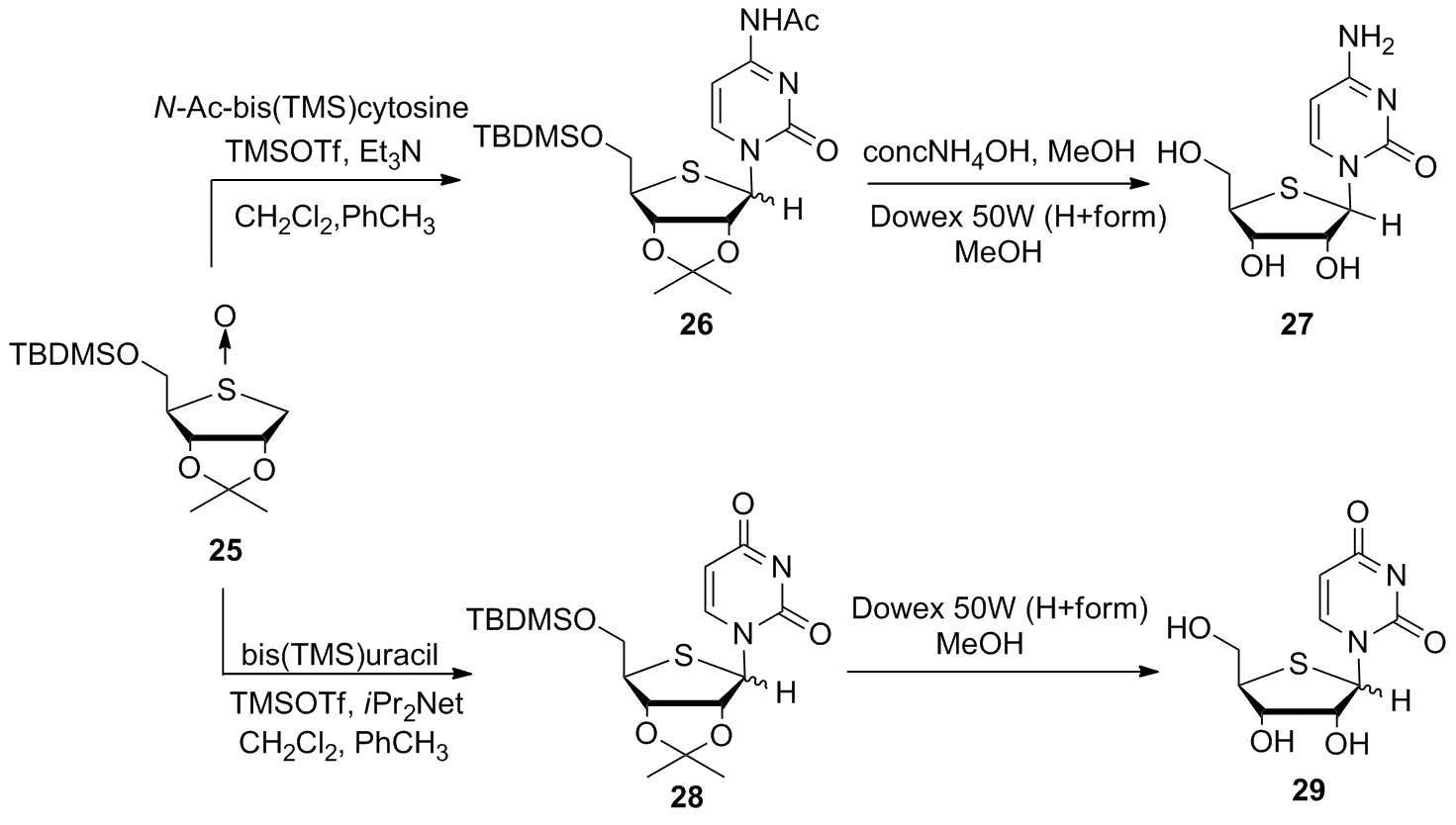
25在三甲基甲硅烷基三氟甲磺酸盐和三乙胺存在下与全硅烷基化的N4-乙酰胞嘧啶反应,生成了以4′-异硫代胞苷衍生物26为主的β-异构体。在氨水/甲醇体系中,通过DOWEX 50W H+阳离子交换树脂实现26的脱保护,以高收率提供纯的β-4′-硫胞苷27。同时,5-O-甲硅烷基化的亚砜25和尿嘧啶以良好的产率得到4′-硫代尿苷衍生物28,具有不可分离的α和β异构体,在酸性条件下甲硅烷基和丙酮化物脱保护,以高收率得到了4′-硫代尿苷(29, Scheme 5)。
Scheme 9
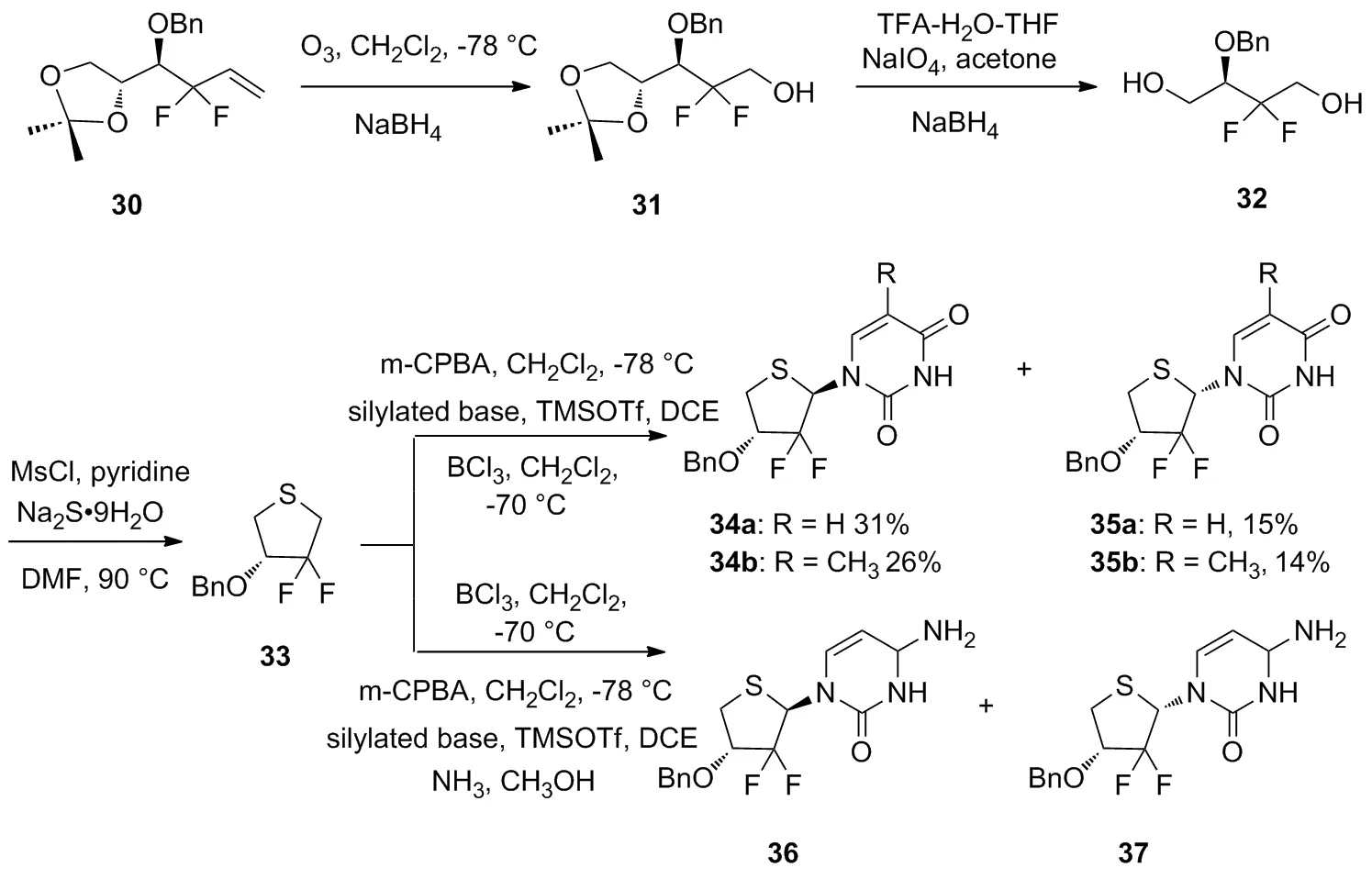
2010年,Zheng等[31]合成D-2′-脱氧-2′,2′-二氟-4′-二氢-4′-硫代核苷。根据报道的方法[32],可以容易地制备二氟化合物30。依次经臭氧氧化,NaBH4还原,将化合物30转化为醇31。再经3步得到目标硫代核苷34,35,36,37(Scheme 6)。
2018年,Yoshimura等[33]设计合成出4′-硫代-2′-脱氧-2′-亚甲基胞苷(4′-硫代DMDC)。其合成由木糖衍生物38开始,制备了噻二环糖39,然后氢化物还原,得到4-硫代阿拉伯糖衍生物40,在伯羟基上保护后得到41,对其氧化后进行Wittig反应,得到亚甲基衍生物,再将其脱苄基化并氧化成相应的亚砜,得到42。接下来,用亚砜42对N4-乙酰胞嘧啶进行Pummerer型硫糖基化得到45。45脱保护得到46及其α-端基异构体(Scheme 7)。
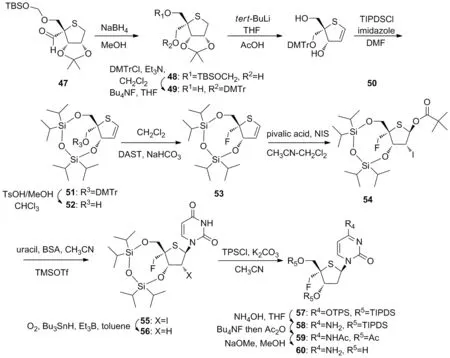
2019年,Haraguchi等[34]设计出4′-C-氟甲基-2′-脱氧-4′-硫胞苷。首先,进行用于4′-C-氟甲基同源物60的糖基供体54的制备。参照文献[35],从D-核糖获得的醛47经一系列反应得到54。接下来,经TMSOTf介导的54与甲硅烷基化尿嘧啶之间的糖苷化反应在分离出72%的情况下得到2′-脱氧-4′-硫尿嘧啶核苷55。锡自由基介导的还原55得到了56。可以从56到57中获得胞嘧啶核苷58。将58转化为乙酸酯59,然后去除59的保护基,得到目标4′-C-氟甲基-2′-脱氧-4′-硫胞苷(60, Scheme 8)。
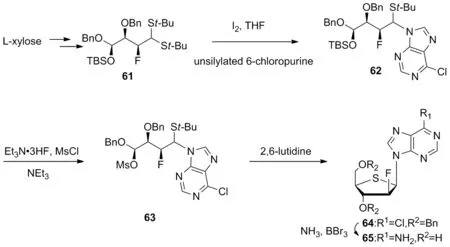
2020年,Cardinal-David等[36]提出了一种无环合成具有腺嘌呤核苷碱基的硫代呋喃糖苷N-糖苷的方法并合成出硫代核苷类似物65。该方法在区域和非对映选择性方面提供了显着的改进。二硫缩醛61可由L-木糖衍生得到,并在碘活化下与未甲硅烷基化的6-氯嘌呤偶联,得到62。使用Et3N·3HF对继发TBS进行脱保护,然后上甲磺酸酯离去基团,得到硫代氨基缩醛63。1′,2′-顺式呋喃呋喃糖苷64只需在2,6-二甲基吡啶中加热即可形成。用氨置换C6-Cl并去除两个苄基保护基团得到了已知的硫代核苷类似物(65, Scheme 9)。
Zhang等[37]在C1和C4位的甲磺酰化66,得到化合物67,随后在DMF中用硫化钠处理等反应,以定量收率得到苯甲酸酯69。用m-CPBA氧化69后与甲硅烷基化的N4-苯甲酰基胞嘧啶缩合,得到区域异构体72与73。用饱和氨的甲醇溶液除去72的苯甲酰基,得到核苷74a和74b。为了证实核苷74a和74b的结构和二氟化噻吩呋喃糖的聚合反应的区域化学,进一步研究了二氟化噻吩磺酰呋喃糖68的Pummerer反应(Scheme 10)。
将68转化为苯氧基硫代糖基衍生物,然后进行后者的自由基脱氧,得到化合物75。用m-CPBA氧化75,然后与甲硅烷基化的N4-苯甲酰胞嘧啶缩合,也得到外消旋核苷77和78。78的脱保护通过氨解作用得到核苷(79, Scheme 11)。
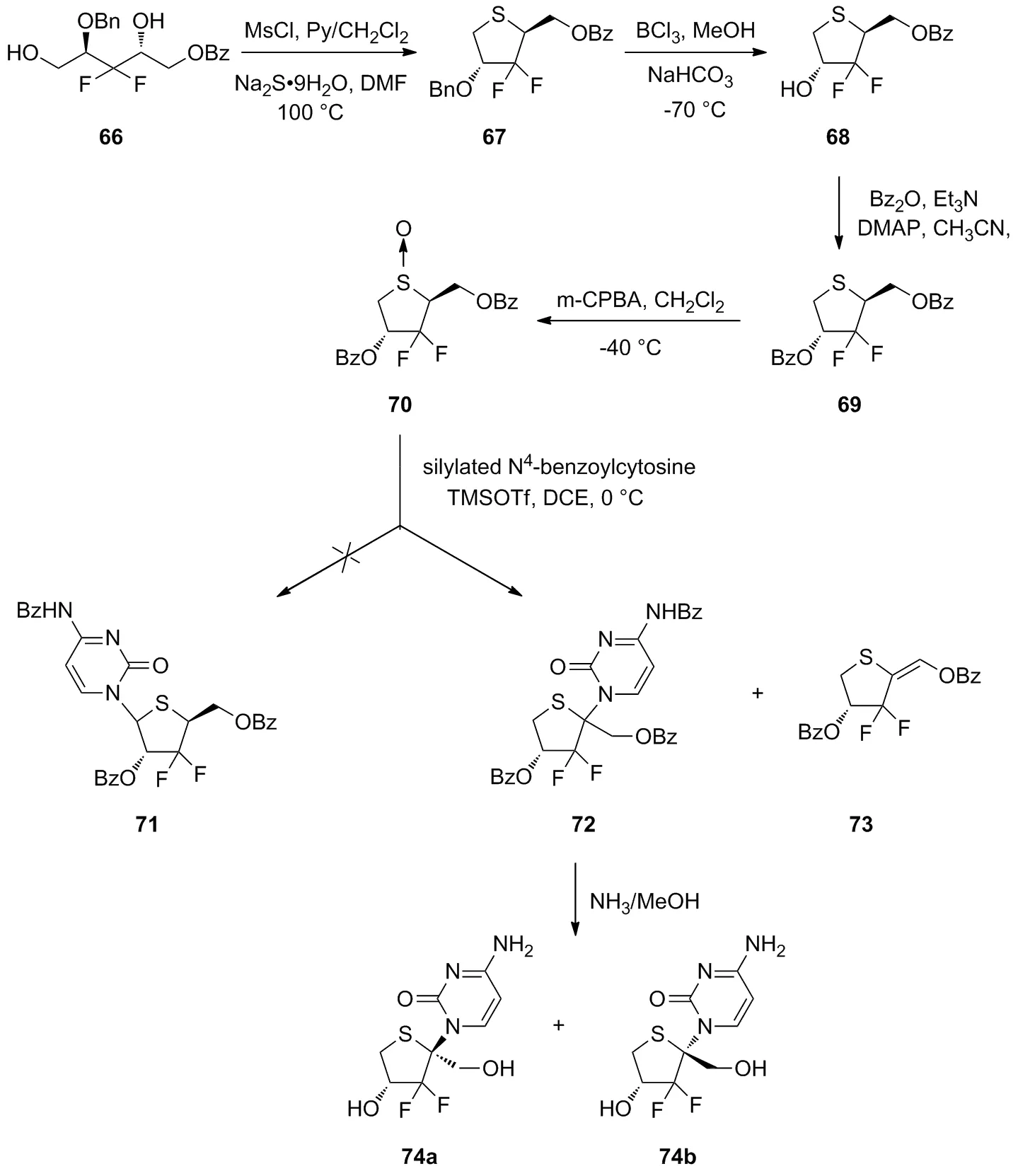
Scheme 10
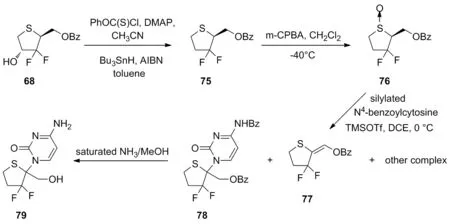
Scheme 11
Lequeux等[38]用黄原酸酯80以三乙基硼烷作为自由基引发剂与多种烯烃反应[39],受保护的醇81a与80在CH2Cl2中反应,得到所需的氟代酯82a后形成硫醇84,在过量的TFA作用下反应,得到内酯85b。用DIBAL-H还原硫代内酯85b,得到获得硫代内酯86,再进一步乙酰化得到87,其准备参与与双三甲基甲硅烷基胸腺嘧啶的Vorbrüggen反应。在新蒸馏的TMSOTf存在下,反应得到硫代核苷类似物(88, Scheme 12)。
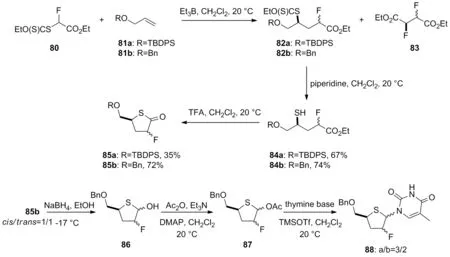
Scheme 12
4′-硫代核苷与4′-氧代核苷具有生物等排关系,并具有固有的优点,例如稳定的糖基键和增加的代谢稳定性。尽管从20世纪90年代以来,鉴于反应步骤的数量和总收率,合成方法得到了很大的改进。但是和与4′-氧代核苷相比,在4-硫糖合成和更高的β-选择性等方面仍有许多问题需要解决。所以对于已经发现的硫代核苷类似物,其合成设计路线应继续完善,用创新性思维减少操作步骤,提高产率,保护环境。对于具有良好生物活性的核苷衍生物,要尽快开发出快捷和经济的合成方法,以降低成本,同时利用计算机模拟的技术对于碳环核苷类似物进行分子设计以及生物活性与化学结构之间关系研究[40]。对于新型硫代核苷类似物的合成与发现仍为研究热点,以扩大核苷类似物数据库,为筛选具有生理活性的核苷类药物打下坚实的基础。
