Different protein expression patterns in rat spinal nerves during wallerian degeneration assessed using isobaric tags for relative and absolute quantitation proteomics profiling
2020-09-18ShuaiWeiXueZhenLiangQianHuWeiShanWangWenJingXuXiaoQingChengJiangPengQuanYiGuoShuYunLiuWenJiangXiaoDingGongHaiHanPingLiuChenHuiShiYuWang
Shuai Wei , Xue-Zhen Liang , Qian Hu , Wei-Shan Wang Wen-Jing Xu Xiao-Qing Cheng Jiang Peng Quan-Yi GuoShu-Yun Liu Wen Jiang Xiao Ding Gong-Hai Han , Ping Liu , Chen-Hui Shi , Yu Wang
1 The First Affiliated Hospital of Medical College, Shihezi University, Shihezi, Xinjiang Uygur Autonomous Region, China 2 Institute of Orthopedics, Chinese PLA General Hospital, Beijing, China 3 Co-Innovation Center of Neuroregeneration, Nantong University, Nantong, Jiangsu Province, China 4 The First Clinical Medical School, Shandong University of Traditional Chinese Medicine, Jinan, Shandong Province, China 5 Kunming Medical University, Kunming, Yunnan Province, China 6 Shanxi Medical University, Taiyuan, Shanxi Province, China
Abstract
Key Words: gene ontology; Kyoto Encyclopedia of Genes and Genomes; isobaric tags for relative and absolute quantitation; motor nerve;proteomics; sensory nerve; spinal nerve; Wallerian degeneration
Introduction
Peripheral nerve injury often leads to limb dysfunction. In China, approximately 700,000 patients are surgically treated for peripheral nerve injury every year (Kearns and Wang,2012). Following peripheral nerve injury, Wallerian degeneration (WD) takes place, and myelin sheath disintegration,axonal degradation, and inflammatory cell infiltration occur at the distal end of the injured axon (Chang et al., 2017;Bittner et al., 2018; Han et al., 2019). While nerve injury causes significant damage to nerve functions, it also plays an important role in peripheral nerve regeneration (Xiong et al., 2016; Caillaud et al., 2019). Wallerian degeneration is the innate immune response of peripheral nerve to injury, rapid removal of degenerated axons and debris of myelin sheath is beneficial to axon regeneration. Nerve injury differences between sensory and motor pathways have been identified in previous work; notably, motor axons withdraw from the skin in a wrong pathway and re-control the appropriate muscle that they belong to (Langley and Anderson, 1904; Ramon y Cajal, 1928). Thus, compared with the skin branches, motor neurons of the femoral nerve can be in a dominant position of muscle branches after transection, even if they have the chance to reach skin branches (Brushart, 1988). There are inherent differences between motor nerve (MN) and sensory nerve (SN) fibers in peripheral nerves, and studies have revealed that these differences play an important role in nerve fiber regeneration during WD (Abdullah et al., 2013).
There are many animal models of peripheral nerve injury,which are most commonly used to study the sciatic and femoral nerves (Cobianchi et al., 2014; Ding et al., 2015; Karegar and Mohammadi, 2015). Some researchers have found that the muscular branch of femoral nerve mainly contains motor nerve fibers, while the cutaneous branch of femoral nerve mainly contains sensory nerve fibers. Because of the purity of muscular and cutaneous nerve branches in femoral nerves,they are often used to study chemotactic regeneration of the peripheral nerve (Zhang et al., 2016; Cai et al., 2017). To improve functional recovery via peripheral nerve regeneration,it is important to have a full understanding of the essential biological differences between SNs and MNs (Raivich and Kreutzberg, 1993). Previous work has revealed that regenerating motor axons express a molecule called polysialylated,which is a form of the neural cell adhesion molecule (Franz et al., 2005, 2008). This suggests that sensory and motor neuron axons have different microenvironment regeneration strategies. However, most peripheral nerves are composed of both SNs and MNs, and it is difficult to find pure SNs or MNs; it is therefore a challenge to examine this hypothesis.To overcome this, most researchers investigate spinal nerves;the anterior and posterior roots of spinal nerves are pure SNs and MNs, respectively, and can therefore be used to exclude the influence of mixed nerve branches, and are therefore ideal for studying different regenerating microenvironments of SNs and MNs.The standard method used for proteomic analysis is two-dimensional electrophoresis followed by MS (mass spec-trometry)-based protein identification (López-Barea and Gómez-Ariza, 2006). This proteomics strategy is a useful method for the comprehensive study of multiple proteins in many tissues (Becker et al., 2008; Lu et al., 2009). Isobaric tags for relative and absolute quantitation (iTRAQ) is a powerful proteomics labeling technique that can be used for quantitating multiple specimens in a single MS run (Unwin et al., 2010). Therefore, the present study used iTRAQ to compare the protein profiles of spinal nerves after injury,and focused on the four following tissue types: pure SNs and MNs in spinal sections from rats before and after injury. The present findings will provide a reference point that will allow future studies to identify differences in WD in MNs and SNs,and better characterize peripheral nerve regeneration.
Materials and methods
Tissue collection
Sixty 8-week-old male adult specific-pathogen-free Sprague-Dawley (SD) rats, weighing 200-250 g, were anesthetized by intraperitoneal injection of moderate sodium pentobarbital (Xingzhi Chemical Plant, Shanghai, China)(30 mg/mL, 30 mg/kg) prior to surgery. After tissue collection, rats received an overdose of sodium pentobarbital to euthanasia (30 mg/mL, 0.6 mL). Twenty rats were used in quantitative real-time polymerase chain reaction (qRTPCR) experiments and forty rats were used in the proteomic analysis. The L2 posterior root nerve and anterior root nerve were transected on the right side. The left side was used as the control. One week later, tissues of the distal stump of the injured L2 posterior root nerve and the proximal stump of the injured L2 anterior root nerve were obtained as the experimental group; the normal L2 posterior root nerve and anterior root nerve were obtained as a control group. There were eight samples in total, which included two from each group. The experimental procedure related to experimental animals were approved by the Ethics Committee of the Chinese PLA General Hospital, China (Approval No. 2016-x9-07) in September 2016. All procedures with experimental animals were performed in accordance with the Guides for the Care and Use of Laboratory Animals. The rats used in this study were raised in a sterile environment and provided by the Laboratory Animal Research Center of The First Clinical Center, Chinese PLA General Hospital (license No.SCXK (Jing) 2016-0002).
Specimen preparation
After the specimens had been obtained, they were quickly placed in cryotubes and stored in liquid nitrogen. Four tissue groups were examined, as follows: tissues of the distal stump of the injured L2 posterior root nerve formed the SN after injury (SI) group; tissues of the normal L2 posterior root nerve formed the normal SN group; tissues of the proximal stump of the injured L2 anterior root nerve formed the MN after injury (MI) group; and tissues of the normal L2 anterior root nerve formed the normal MN group. All samples were dissolved by vortex mixing, adequately homogenized in 1 mL of lysis buffer, and then disposed by the sonicate (Xu et al., 2017) at 120 W for 60 seconds, which was conducted on an ice-water mixture. The supernatant was collected and stored after ultrasonic extraction and centrifuged for 20 minutes. Then, the concentrations of proteins were measured using the Bradford method (Bradford, 1976).
Isobaric tags for relative and absolute quantitation labeling and liquid chromatography-mass spectrometer/mass spectrometer analysis
On the basis of the specification requirements, compound samples were extracted, then labeled using the iTRAQ Reagent-8plex Multiplex Kit (PN:4390812; AB SCIEX, Framingham, MA, USA). The SN samples were labeled with iTRAQ tags 113 and 114, and the SI samples were labeled with tags 115 and 116; the MN samples were labeled with iTRAQ tags 117 and 118, and the MI samples were labeled with tags 119 and 121. Later, the total peptides labeled with iTRAQ tags were mixed at equal proportions, then decomposed with high pH C18 chromatography, and 12 fractions were obtained. Finally, the total fractions were analyzed by liquid chromatography-MS and separated by an AB SCIEX system with a buffer composed of buffer A (0.1% formic acid and 5% acetonitrile), buffer B (95% acetonitrile and 0.1%formic acid), and a loading buffer (0.1% formic acid and 3%acetonitrile). The MS equipment comprised an AB SCIEX analytical column, a new objective needle, and a Chromxp Trap Column.
Analysis of differentially expressed proteins
To identify the main functional pathways of the differentially expressed proteins (DEPs) in injured nerves, the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes(KEGG) pathway-enrichment analyses were performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) 6.8 (https://david.ncifcrf.gov/). The statistical threshold was set as P < 0.05 with fold change (FC)≥ 1.5.
Protein-protein interaction networks construction and analysis
Based on the differentially expressed proteins identified in the previous step, we constructed a protein-protein interaction (PPI) network. For this, the search tool for the retrieval of interacting genes (STRING, version 10.5, https://stringdb.org/) database was used; DEP pairs with a combined score > 0.4 were identified. Only connected nodes were retained, and Cytoscape (version 3.6.1, https://cytoscape.org/) was used to create a visualization of the network. The topological properties of every connected node in the PPI network were assessed using Network Analyzer, which revealed the topological significance of each connected node.The greater the quantitative value of the node, the greater its importance in the network.
Based on a previous study by Zhang et al. (2013), signifi- cant targets were defined as nodes, of which the degrees were more than twice that of the median degree in all nodes. To explore the molecular complexes and the specific regulatory relationships between proteins in the PPI network, module analysis of the network was conducted using MCODE1.5.1(https://cytoscape.org/), which is a plugin for Cytoscape(Halary et al., 2010) that calculates node information such as neighbors and density for each node in the network graph. It then builds functional modules for clustering using K-Core analysis. Data parameters were set with applicable thresholds of K-Core > 5.
Gene Ontology and Kyoto Encyclopedia of Genes and Genomes analyses
The DAVID database provides a comprehensive set of functional annotation, visualization, and integrated discovery tools (Huang da et al., 2009) to allow researchers to understand the biological implications of a large list of genes. It can be used to perform typical batch annotation and GO terminology-rich analyses to identify the related term of GO that have a close relationship with the gene list. The actual edition includes more than 40 categories, including GO terminology, PPI, general sequence characteristics, homology,gene function summaries, gene organization expression, and literature. GO enrichment and KEGG analyses of the above key differential genes were performed using the DA-VID 6.8 online analysis tool, and the R language ggplot2 installation package was used to draw bubble charts.
Quantitative real-time polymerase chain reaction
The total RNA was extracted from nerve tissues with TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA).Every RNA sample was reverse transcribed into cDNA with a reverse transcription kit (TaKaRa, Tokyo, Japan) to detect mRNA, then qRT-PCR was used to quantify mRNAs levels.Glyceraldehyde-3-phosphate dehydrogenase was set as an internal reference. Detailed information about all primers is shown in Table 1.
Statistical analysis
The data are shown as the mean ± SD. Between-group differences were analyzed using a two-sample t-test and this analysis was performed using SPSS 22.0 (IBM, Armonk, NY, USA).The qRT-PCR results were shown by a histogram and created using GraphPad Prism 6.0 (GraphPad Software Inc., San Diego, CA, USA). Statistical significance was set at P < 0.05.
Results
Identification of proteins different with expression in the four sample types
A total of 626 proteins that were differentially expressed in both SN and SI groups were identified, of which 368 were upregulated and 258 were downregulated in response to the ratio of SN/SI. A total of 637 proteins that were differentially expressed in both MN and MI groups were identified, of which 372 were upregulated and 265 were downregulated in response to the ratio of MN/MI. All different proteins identified in the SN, SI, MN, and MI groups that were up- and down-regulated are shown in a heatmap (Additional Figure 1).

Table 1 Sequences of primers used for quantitative real-time polymerase chain reaction
Protein-protein interaction networks
By confluence different proteins using a composite score > 0.4,we constructed a network of PPI for the SN and SI groups using the STRING database. The network included 581 nodes (different proteins) and 4012 edges, which ac-counted for 92.81% of all proteins. For the MN and MI groups, the PPI network included 492 nodes (different proteins) and 3450 edges, which accounted for 77.24% of all proteins. The PPI networks were visualized in Cytoscape, through which relevant nodes were chosen for thorough analysis, and several irrelevant nodes were discarded. The resulting refined networks contained 358 nodes and 2446 edges for the SN and SI groups, and 443 nodes and 3450 edges for MN and MI groups. To analyze and process the large networks conveniently, they were extracted from the whole network. Based on a related study by Zhang et al. (2013), the specific nodes of all nodes of the SN and SI groups and 443 nodes of MN and MI groups were identified, of which the degrees were more than twice the median degree in all nodes. Thus, a network of candidate targets was constructed for SN and SI groups that had 87 nodes and 1067 edges, including 42 upregulated and 45 downregulated proteins (Figure 1). The network of candidate targets for the MN and MI groups had 131 nodes and 1768 edges, including 78 upregulated and 53 downregulated proteins (Figure 2).
Biological networks consist of various functional parts,which may be involved in biological processes of acquaintance. Hence, we created a new perspective on the biological function of complex networks. Using the MCODE analysis with a K-Core > 5, two were modules extracted from the PPI network. For the SN and SI network, one identified cluster included 30 nodes and 234 edges (cluster rank 1; K-Core 16.138), and the other included 26 nodes and 108 edges(cluster rank 2; K-Core 8.640; Figure 3A & B). For the MN and MI network, one identified cluster consisted of 24 nodes and 202 edges (cluster rank 1; K-Core 17.565), and the other consisted of 26 nodes and 165 edges (cluster rank 2; K-Core 13.200; Figure 3C & D).
Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway analysis of functional patters of different genes
To reveal more specific functional patterns of these genes correspond to differently expressed proteins, 286 biological process items, 167 cellular component items, 124 molecular function items, and 57 KEGG signaling pathway items of SN and SI groups were obtained by introducing key differentially expressed genes into the DAVID online data tool for functional enrichment and pathway analysis; the top 10 significant P-values are presented as a bubble diagram (Figure 4).For the MN and MI groups, 65 biological process items, 42 cellular component items, 26 molecular function items, and 57 KEGG signaling pathway items were obtained; the top 10 P-values are presented as a bubble diagram (Figure 5). The results were divided into the two following parts: molecular functions/cellular components/biological processes and the KEGG pathway. In the SN and SI network, differentially expressed genes linked to biological processes were mainly related to cell-cell adhesion, carbohydrate metabolic processes, cell adhesion, and aging (Figure 4A); cellular components were mainly related to extracellular exosomes, myelin sheath,membrane, and cytoplasm (Figure 4B); molecular functions were mainly related to protein binding, cadherin binding involved in cell-cell adhesion, poly(A) RNA binding, and actin filament binding (Figure 4C); and the KEGG pathway was mainly related to antibiotic biosynthesis, carbon metabolism,amino acid biosynthesis, and protein processing in the endoplasmic reticulum (ER; Figure 4D). For the MN and MI network, the identified cellular components were mainly related to the presence of extracellular exosomes and myelin sheath,focal adhesion, and cytosol (Figure 5A); molecular functions were mainly related to poly(A) RNA binding, adenosine triphosphate (ATP) binding, protein disulfide isomerase activity, and GTPase activity (Figure 5B); biological processes were mainly related to glycolytic process, cell redox homeostasis, protein folding, and citrate metabolic processes (Figure 5C); and the KEGG pathway was mainly related to carbon metabolism, antibiotic biosynthesis, glycolysis/gluconeogenesis, and amino acid biosynthesis (Figure 5D).
Quantitative real-time polymerase chain reaction verified the expressions of key genes
The expressions of key genes in the SN, MN, SI, and MI tissues were measured using qRT-PCR. The expression of Akt1 was upregulated in SI samples compared with SN samples(MI vs. MN, P = 0.0481; SI vs. SN, P < 0.0001). However, the expression of Snap25 (MI vs. MN, P < 0.0001; SI vs. SN, P <0.0001), Kcna1 (MI vs. MN, P < 0.0001; SI vs. SN, P = 0.0004),Kif5b (MI vs. MN, P = 0.001; SI vs. SN, P = 0.0001), Cntnap1(MI vs. MN, P = 0.0131; SI vs. SN, P = 0.0553), and Palm (MI vs. MN, P < 0.0001; SI vs. SN, P < 0.0001) was lower in SI tissues than SN (Figure 6). The same genes were differentially expressed between MN and MI, with the same expression patterns. These results suggest that these genes may be involved in the injury process of sensory and motor nerves.
Discussion
Peripheral nerve injury can have adverse effects which can prolong the recovery time of injured nerves and affect the function of limbs, when nerve fibers become wrongly connected. Although some progress has been made toward treating peripheral nerve injury, such as tension reduction suture of nerve stumps, autogenous nerve transplantation,and artificial nerve conduits, the clinical outcome remains unsatisfactory. Hence, it is urgent to clarify the mechanism of nerve injury to facilitate further treatment strategies for peripheral nerve injury. The present study revealed different protein expressions in the rat spinal nerve during WD using iTRAQ proteomics profiling and identified DEPs that could reveal differences and similarities between SNs and MNs during WD. The DEPs were correlated with a series of biological processes and pathways; the following discussion focuses on several types of DEPs found in SNs and MNs before and after injury.
Isobaric tags for relative and absolute quantitation analysis of the unique peptide
A unique peptide is one that is only present in one protein; detection of a unique peptide indicates the presence of the corresponding protein in a sample. The distribution of unique peptides shown a two-coordinate distribution curve of the number of unique peptides found in all proteins screened in this experiment (Additional Figure 2). A total of 3391 proteins contained at least two unique peptides,accounting for 78.64% of the total proteins. Each MS has its own measurement range and, thus, the identified peptides have certain length limitations. Therefore, if the peptide content detected by the MS used in the present study was too low or too high, the protease selection may have been inappropriate. The average polypeptide length was 13.6, which is within the reasonable range of peptide lengths (Additional Figure 3).
Key biological processes during wallerian degeneration in sensory nerves
A total of 626 DEPs were screened in both SN and SI groups,of which 368 were upregulated and 258 were downregulated in response to WD. Results from the biological processes analysis, which identified the certain biological processes involved in the WD of SNs, along with those of the GO enrichment analysis suggested that these key genes are involved in cell-cell adhesion, carbohydrate metabolic processes, cell adhesion, aging, and protein folding.
Cell-cell adhesion and contact constitute fundamental physiological processes of multicellular organisms. Tissues are formed by different types of cells, and these cells must remain in physical contact with each other for structural and functional communications (Murakawa and Togashi, 2015).In the peripheral nervous system, the interactions between Schwann cells and axons also appear to be influenced by the deformation of Schwann cells in the microenvironment of injured and developing nerves (Namgung, 2014). In regenerating nerves, the guidance of the growth cone, axonal elongation, and myelination are correlated with the presence of Schwann cell and axon interaction (Pereira et al., 2012).The carbohydrate metabolic process could provide energy for many biological processes in tissues and could underlie different types of physiological activity, including WD.Many genes are involved in aging and, in the present study,Hspa8, which is the gene for the heat shock protein 70, had a higher expression level in SNs during WD compared to the normal nerve. Because of after its expression induced by ischemic stress, heat shock protein 70 may play a key role in protecting cells under mild stress conditions (Miller et al.,1991). Additionally, the unfolded or misfolded proteins in the ER accumulate as a result of multiple types of adverse stimulation, such as hypoxia, nutrient deprivation, and viral infection. Afterwards, the stress condition of the ER will be in turn triggered, and an evolutionarily conserved unfolded protein response will occur (Ron and Walter, 2007). In an animal model of optic nerve crush, in which most retinal ganglion cells are dead, Hu et al. (2012) demonstrated that the unfolded protein response was activated by axon injury in distinct pathways. In the present study, many genes were involved in the protein folding of biological processes,including Cct3, Hspa8, Hspa9, and Hspa4l. These findings may indicate that unfolded or misfolded proteins appear in injured nerve tissues during WD.
Key biological processes during wallerian degeneration in motor nerves
A total of 637 DEPs were screened in both MN and MI groups, of which 372 were upregulated and 265 were down-regulated. The GO enrichment analysis suggested that these genes take part in the glycolytic process, cell redox ho-meostasis, and protein folding.
Much like the biological process of carbohydrate metabolism, the glycolytic process produces ATP for normal or abnormal physiological activities, including WD. In a study of the genesis of neuropathic pain, Lim et al. (2015) found increased oxygen consumption and lactate levels in the endoneurium in a mouse model of peripheral nerve injury, which suggests that the ATP levels in cells are maintained by anaerobic glycolysis. The changes of redox in cells plays a key part in the regulation of cell growth and aging, and have been reported to be correlated with a variety of neurodegenerative diseases (Kwon et al., 2003). In addition, because numerous signaling pathways within cells are sensitive to hypoxia conditions, altered intracellular redox plays an important role in the cell , such as cell homeostasis, cell proliferation and cell apoptosis (Chiu and Dawes, 2012). After injury, nerve tissues are under hypoxic conditions due to inflammatory reactions and ischemia. To assess whether injured nerves were hypoxic, Cattin et al. (2015) injected pimonidazole hydrochloride to rats (Xu et al., 2017) which can translate into immunofluorescent protein adducts detectabled in the conditions of hypoxic. The results indicated that there are numerous hypoxic cells before vascularization. It is known that the levels of reactive oxygen species in the brain increase with increasing blast pressure and injury , and immunohistochemical analysis of the brain sections analyzed demonstrated astrocytosis and cell apoptosis, confirming sustained neuronal injury response (Kabu et al., 2015). In the present study, many upregulated proteins were related to cell redox homeostasis, including prdx1, prdx2, prdx6, p4hb, and gpx1.The phosphorylation of prdx1, a major H2O2scavenger and signaling regulator, is related to the accumulation of localized H2O2that is involved in cell signaling (Woo et al., 2010).Additionally, similarly to the biological process of protein folding in SNs, there were many highly expressed proteins in

Figure 1 Identification of significantly different proteins in the SN and SI networks.
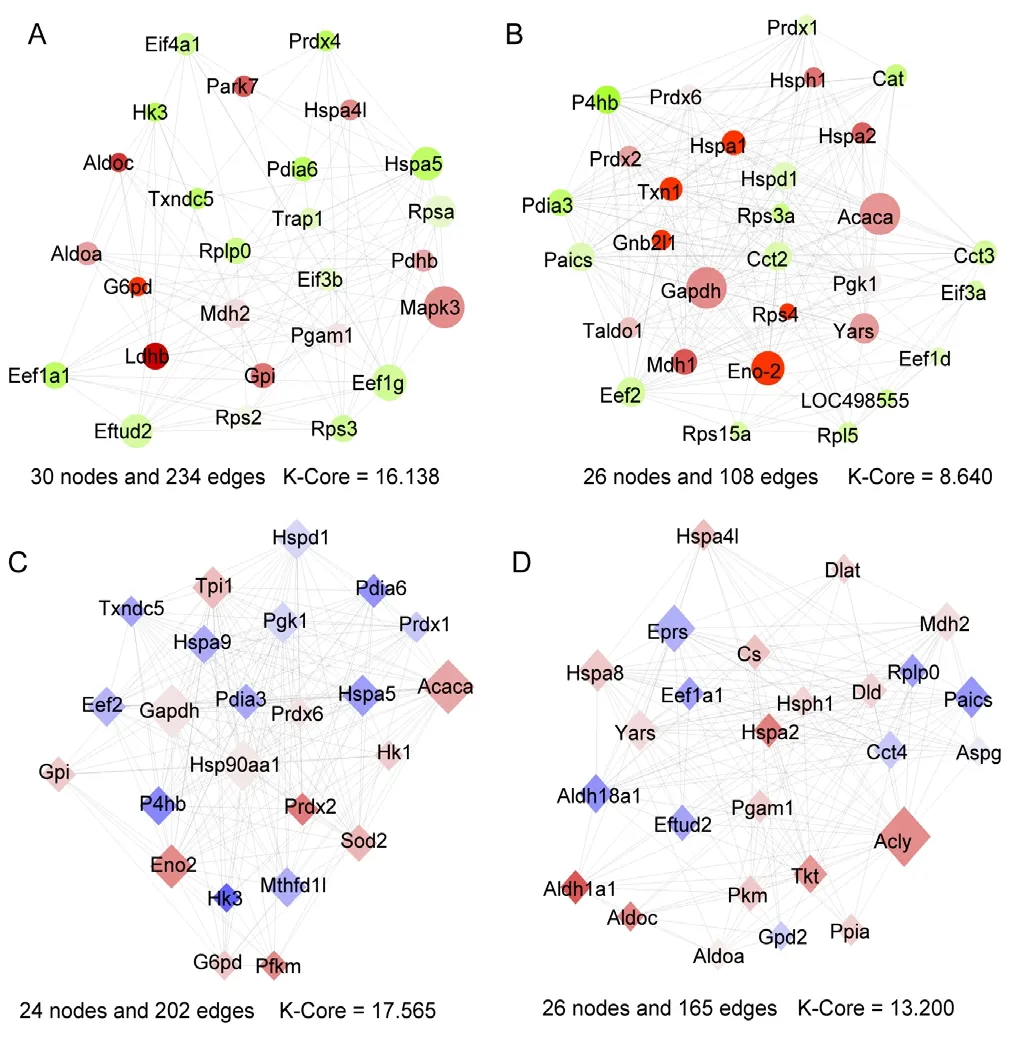
Figure 3 Sub-module analysis of the most significant nodules extracted from the giant network.

Figure 2 Identification of significantly different proteins in the MN and MI networks.
MNs, including Cct4, Hsp90aa1, Pdia3, and Pdia6. Protein folding is commonly enriched between sensory and MNs and may be a significant biological process during WD.
Common characteristics between sensory and motor nerves during wallerian degeneration in Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway analyses
In the GO ontology assessments of the cellular components and molecular function, the SNs and MNs appeared to have many common characteristics. The GO enrichment analysis indicated that these common genes were associated with the presence of extracellular exosomes, myelin sheath, and focal adhesion in the cellular component ontology between sensory and MNs. After nerve injury, WD begins with the degradation of the axons and myelin sheath. The most obvious characteristic of Schwann cell demyelination is that the myelin sheath decomposes into small ovoid-like structures,and the expression of the myelin gene is downregulated at the molecular level (Jessen and Mirsky, 2008). The present study observed lower expression levels of genes related to the myelin sheath in sensory and MNs, which indicates that degradation of the myelin sheath is a basic feature of WD.
Schwann cells in the peripheral nervous system release exosomes (Lopez-Verrilli and Court, 2012) and may play significant roles in neurodevelopment, neurodegeneration,and neuroprotection (Lai and Breakefield, 2012; Kalani et al.,2014). Peripheral nerve axons can internalize the exosomes released by Schwann cells, which have a positive impact on neurite outgrowth in vitro (Lopez-Verrilli et al., 2013). The present study also confirmed the existence of extracellular exosomes during WD, which implies that Schwann cell exosomes and their genetic cargo likely represent a vital component in the processes of WD and nerve regeneration in both sensory and MNs.
It is a little-known process that growth cone adhesion toextracellular matrix proteins at specific sites called point contacts (Renaudin et al., 1999). Point contacts come into being by growth cones in the microenvironment of nerve regeneration, which are composed of several key focal adhesion-related molecules, such as focal adhesion kinase (FAK),integrins, paxillin, and vinculin (Robles and Gomez, 2006).(Myers and Gomez, 2011) found that the control of adhesion mediated by FAK is essential for axon steering, which shows that the regulation of point contacts play an important role in the process of axon guidance. The present study also identified additional genes related to focal adhesion in sensory and MNs, including Myh9, Calr, Rps3, and Actr3. Baeyens et al. (2011) found that non-physiological metabolism of EBP50 harms myosin IIa fibers that are encoded by the Myh9 gene, which results in decrease of focal adhesion formation.During cell migration, similarly to axon elongation, the assembly and disassembly of integrin-based focal adhesions is necessary, and can connect the actin cytoskeleton (encoded by the Actr3 gene) to the extracellular matrix (Ridley et al.,2003). These findings suggest that regenerating axons may exist in the later stages of WD.

Figure 4 Enrichment analysis of 87 genes from the SN and SI networks.
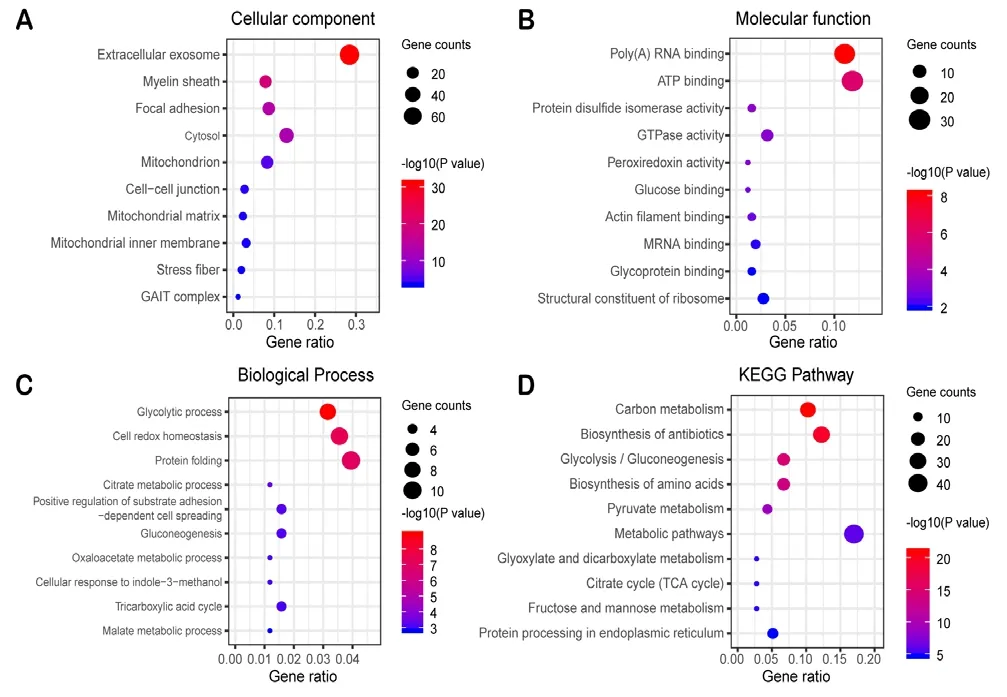
Figure 5 Enrichment analysis of 131 genes from the MN and MI network.

Figure 6 The expression of key genes in SN, MN, SI and MI tissues detected by quantitative real-time polymerase chain reaction.
Additionally, our KEGG enrichment analysis indicated that these common genes participated in carbon metabolism, metabolic pathways, protein processing in the ER biosynthesis of amino acids, and the biosynthesis of antibiotics during WD in both sensory and MNs. It is widely accepted that disturbances in nucleotide metabolism, ATP synthesis,and related purinergic signaling pathways due to injury trigger a set of cellular responses that protect the cell from virus invasion and/or homeostasis disorder (Naviaux, 2014). The type of cell damage has been labeled using a variety of terms,including the stress response of ER (Liu et al., 2008), the misfolded and unfolded protein response (Lee and Glimcher,2009), and the oxidative stress response (Lushchak, 2011).
Key proteins in wallerian degeneration in both sensory and motor nerves
Based on these results of GO and KEGG analysis, several genes were chosen for qRT-PCR validation, as follows: Akt1,Snap25, Kcna1, Kif5b, Cntnap1, and Palm. Importantly, the qRT-PCR results were consistent with the regulation of protein expression in iTRAQ. In the early stages of nerve injury,Schwann cells in both the proximal stump and throughout the nerve downstream of the injury dedifferentiate to progenitor-like cells, which proliferate and orchestrate an inflammatory response that clears the debris and remodels the environment. At the same time, WD occurs, and myelin sheath disintegration, axonal degradation, and inflammatory cell infiltration take place at the distal end of the injured axon (Conforti et al., 2014). There were no differences in expression in the chosen genes between MI and SI samples.Akt1, which is associated with the cellular response to DNA damage stimuli and hypoxia, has been reported to be upregulated in both the posterior and anterior roots of the spinal nerve after injury (Luo et al., 2010). Snap25, Kcna1, Kif5b,Cntnap1, and Palm, which are associated with the biological processes of axonogenesis, neuron differentiation, maintenance of membrane potential, nerve signaling, and axon guidance, have been reported to be downregulated in both the posterior and anterior roots of the spinal nerve after injury (Poliak et al., 1999; Sidor-Kaczmarek et al., 2004; Kutzleb et al., 2007; Ishida et al., 2012; Sun et al., 2013).
While different protein expression patterns in rat spinal nerves have been studied previously (He et al., 2012), protein expression patterns before and after WD have rarely been investigated. Because the spinal cord belongs to the central nervous system and the spinal nerve belongs to the peripheral nerve, which is very close to the central nervous system in anatomy. Thus, the spinal nerve may be disturbed by the central nervous system, which is one limitation of this experiment. It’s still not totally clear what this limitation is, exactly,or how it might have affected the results. WD is an important component of the regeneration process after peripheral nerve injury. While it promotes nerve regeneration, it can also lead to the impairment of nerve function. However,the molecular mechanisms underlying WD are still unclear.Therefore, on the basis of the results of this study, we are in the process of further exploring the aspects of functional and pathway for the related proteins.
In summary, the present study screened DEPs that may potentially reveal differences and similarities between SNs and MNs during WD. The present findings could provide a reference point for a future investigation into the differences between sensory and motor nerves in Wallerian degeneration and the characteristics of peripheral nerve regeneration.
Acknowledgments:We are very grateful to Xun Sun, MD, from Department of of Orthopedics, Tianjin Hospital, China, for his selfless academic guidance in the process of polishing up the manuscript and Qian Hu, Master, from Shihezi University, China, for her great moral encouragement.
Author contributions:Study design and paper drafting: SW, XZL, QH;experimental guidance and manuscript revision: WSW, WJX, JP, QYG,SYL; data collection and analysis: XQC, WJ, XD, GHH, PL; study methods selection of and technical support: CHS, YW. All authors read and approved the final manuscript.
Conflicts of interest:The authors declare that there is no conflict of interests regarding the publication of this paper.
Financial support:This study was supported by National Key Research & Development Program of China, No. 2016YFC11011601,2017YFA0104701, the Youth Cultivation Project of Military Medical Science, China, No. 15QNP091 (to YW), and People's Liberation Army Youth Training Project for Medical Science of China, No. 16QNP144 (to YW). The funding bodies played no role in the study design, in the collection, analysis and interpretation of data, in the writing of the paper, or in the decision to submit the paper for publication.
Institutional review board statement:This study was approved by the Institutional Animal Care and Use Committee of the Chinese PLA General Hospital (Approval No. 2016-x9-07) in September 2016.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak,and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:
Additional Figure 1: The totally different proteins in the SN, SI, MN, and MI groups that were up- and downregulated are shown in a heatmap.
Additional Figure 2:The distribution of unique peptides shows a two-coordinate distribution curve of the number of unique peptides found in all proteins screened in this experiment.
Additional Figure 3:The peptide length distribution shows the length distribution of the peptides detected by this mass spectrometer.
杂志排行

中国神经再生研究(英文版)的其它文章
- Ethanol extract from Gynostemma pentaphyllum ameliorates dopaminergic neuronal cell death in transgenic mice expressing mutant A53T human alpha-synuclein
- Peripheral nerve injury induced changes in the spinal cord and strategies to counteract/enhance the changes to promote nerve regeneration
- Genetic targeting of astrocytes to combat neurodegenerative disease
- Pathological significance of tRNA-derived small RNAs in neurological disorders
- Applications of advanced signal processing and machine learning in the neonatal hypoxic-ischemic electroencephalography
- Protective effect of hydrogen sulfide on oxidative stress-induced neurodegenerative diseases