Action of trichostatin A on Alzheimer's disease-like pathological changes in SH-SY5Y neuroblastoma cells
2020-09-18LiHuaLiWenNaPengYuDengJingJingLiXiangRongTian
Li-Hua Li , Wen-Na Peng, Yu Deng Jing-Jing Li Xiang-Rong Tian
1 College of Medicine, Jishou University, Jishou, Hunan Province, China 2 Department of Rehabilitation, Second Xiangya Hospital, Changsha, Hunan Province, China 3 College of Biology and Environmental Science, Jishou University, Jishou, Hunan Province, China
Abstract
Key Words: Alzheimer's disease; amyloid-β peptide; autophagy; Keap1 signal; neurocytotoxicity; oxidative stress damage; SH-SY5Y cells; total antioxidant capacity; transcription factor Nrf2; TSA
Introduction
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by memory impairment and behavioral disorders (Dodich et al., 2016; Lin et al., 2018; Mokhtar et al.,2018; Zhang et al., 2019), and is pathologically characterized by the presence of extracellular amyloid plaques, intracellular neurofibrillary tangles, and synapse loss (Hong et al.,2016; Zhao, 2016; Filadi and Pizzo, 2019). Understanding the mechanism of AD is crucial for preventing and treating the disease. Mutations in amyloid precursor protein are associated with increased production of amyloid β-peptide (Aβ)(Waring and Rosenberg, 2008; Selkoe and Hardy, 2016).Aβ disturbs neuronal metabolism (Kuhla et al., 2004; Campos-Pena et al., 2017) and Aβ in senile plaques can amplify microglial activation by a coexisting submaximal inflammatory stimulus (Verbeeck et al., 2017). Aβ-mediated oxidative stress is considered to cause neuronal damage and to be a major factor in AD pathogenesis (Hardy and Selkoe, 2002;Shioi et al., 2007; Jiang et al., 2016). It is well documented that Aβ induces some of the symptoms of AD (Selkoe, 1994;Hardy and Selkoe, 2002; Wang and Liu, 2012; Bruggink et al., 2013; Tönnies and Trushina, 2017). Aβ protein fragment 25-35 (Aβ25-35) is widely used to establish in vitro cell models of AD (Kaminsky et al., 2010; Wang and Liu, 2012; Chang and Teng, 2015). Recent therapeutic research has focused on developing amyloid plaque-specific antibodies and antioxidants to protect against Aβ-mediated oxidative stress (Hardy and Selkoe, 2002; Youn et al., 2014).
The Kelch-like ECH-associated protein 1 (Keap1) and nuclear factor erythroid 2-related factor 2 (Nrf2) pathway mainly regulates the oxidative and xenobiotic stresses of cellular defense responses (Zhang, 2006; Noel et al., 2015; Galiè et al., 2018). Under homeostatic conditions in the absence of stress, Keap1 binds to Nrf2 to constitutively suppress Nrf2 activity (Itoh et al., 1999; Hanada et al., 2012), resulting in Nrf2 accumulation (Itoh et al., 1999; Magesh et al., 2012).Consequently, Nrf2 translocates to the nucleus and activates transcription of its target genes (Rangasamy et al., 2004;Zhang, 2006; Moon and Giaccia, 2015; Tebay et al., 2015),including phase II detoxification enzymes, antioxidant proteins, xenobiotic metabolism enzymes and proteasome subunits (Nguyen et al., 2003; Rangasamy et al., 2004; Zhang,2006; Hanada et al., 2012), which are involved in regeneration of small molecule antioxidants (Dinkova-Kostova and Talalay, 2008). The activity of Nrf2 is inhibited in AD (Sykiotis and Bohmann, 2010), and Nrf2 plays a protective role by regulating downstream gene transcription (Ramsey et al.,2007; Kanninen et al., 2009; Chapple et al., 2017). Numerous studies show that activating Nrf2 protects against toxicity and prevents neuronal lesions in mammalian cells and mouse models of AD (Kanninen et al., 2008; Kanninen et al., 2009; Yang et al., 2009; Sun et al., 2017; Ishii et al., 2019).Keap1 and glycogen synthase kinase-3 are well known as Nrf2 inhibitors (Zhang, 2006; Rada et al., 2011; Abed et al.,2015). Inhibition of glycogen synthase kinase-3 can protect against toxicity induced by the Aβ42 peptide (Sofola et al.,2010; Konstantinopoulos et al., 2011; Abed et al., 2015; Kerr et al., 2017). The Nrf2-antioxidant response element (ARE)pathway can be activated through Keap1 inhibition by a direct Keap1 inhibitor (Bertrand et al., 2015), siRNA knockdown of Keap1 (Youn et al., 2014), and suberoylanili dehydroxamic acid treatment (Eades et al., 2011). Given these observations, the Keap1-Nrf2 pathway is a promising target for AD prevention and treatment (Eades et al., 2011; Abed et al., 2015; Kerr et al., 2017).
Histone deacetylase inhibitors are promising pharmacological agents for AD treatment (Xu et al., 2011; Seo et al.,2013; Yang et al., 2014a). Trichostatin A (TSA), a histone deacetylase inhibitor, improves memory and learning abilities in mouse models of AD (Yang et al., 2014a, b). However, the underlying mechanism of its action is not well understood.TSA promots gelsolin expression in a transgenic mouse model of AD (Yang et al., 2014a) and prevents the formation of new amyloid deposits (Yang et al., 2014b). In addition,TSA inhibited calcium-induced SH-SY5Y cell toxicity (Seo et al., 2013) and combated cognitive impairment associated with metabolic syndrome (Sharma et al., 2015). Suberoylanili dehydroxamic acid, a histone deacetylase inhibitor, can down-regulate Keap1 and activate Nrf2 target genes (Eades et al., 2011). Nevertheless, the possible relationship between TSA and the Keap1-Nrf2 signaling pathway has not been studied.
In this study, the human neuroblastoma cell line, SH-SY5Y,was treated with Aβ25-35to establish an in vitro cell model of AD. The therapeutic effects of TSA and its relationship with the Keap1-Nrf2 pathway were examined. We demonstrate that TSA attenuated Aβ-induced oxidative stress injury and autophagy, and increased total antioxidant capacity by inhibiting Keap1 expression and promoting Nrf2 to enter the nucleus to activate downstream signals. These findings support the use of TSA as an efficient therapy for AD.
Materials and Methods
Cell culture and treatment
The human neuroblastoma cell line, SH-SY5Y, was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). SH-SY5Y cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Waltham, MA,USA) supplemented with non-essential amino acids, 10%fetal bovine serum (Gibco) and 1% penicillin-streptomycin(Gibco) at 37°C, in a 5% CO2atmosphere. To establish an in vitro cell model of AD, cells were treated with 25 μM Aβ25-35(A2201, Sigma, St Louis, MO, USA) for 24 hours. SH-SY5Y cells were incubated in a series of H2O2Concentrations(200, 400, and 600 μM; Yamin Biomedical, Shanghai, China)for 24 hours to induce oxidative injury. To study the effect of TSA (V900931, Sigma), in vitro cell models of AD were pre-incubated with various concentrations of TSA (10, 20,40, 60, 80, and 100 ng/mL) for the indicated times (24, 48,and 72 hours). An inverted microscope with a 10× objective(Leica, Wetzlar, Germany) was used to observe cells.
MTT assay for cell viability
To study the effect of TSA (V900931, Sigma) on cell viability we used 4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay. In vitro AD model cells(20,000 cells/well) in 96-well plates were pre-incubated with various concentrations of TSA (10, 20, 40, 60, 80, and 100ng/mL) for the indicated times (24, 48, 72 hours). Then 10 μL MTT solution (5 mg/mL) was added to each well. After incubation for 4 hours at 37°C, the culture medium was removed and formazan crystals were solubilized with 100 μL dimethyl sulfoxide. Absorbance was measured at 570 nm with a microplate reader (Bio-Rad Laboratories, Hercules,CA, USA). Cell viability was determined as a percentage of the optical density value relative to that of the control group.
western blot analysis
SH-SY5Y cells were incubated for 24 hours with TSA (60 ng/mL) and then whole-cell, cytoplasmic, and nuclear extracts were prepared. Samples were separated by sodium dodecyl sulfate 10% polyacrylamide gel electrophoresis and then transferred onto polyvinylidene fluoride membranes. After blocking with 5% bovine serum albumin (Gibco) in Tris-buff-ered saline at 37°C for 2 hours, membranes were incubated overnight at 4°C with primary antibody. Primary mouse monoclonal antibodies against Keap1 (1:500; E-20, Santa Cruz Biotechnology, Dallas, TX, USA), Nrf2 (1:800; H-300,Santa Cruz Biotechnology), Lamin B (1:500; sc-56144, Santa Cruz Biotechnology), GAPDH (1:2000; 2118S, Cell Signaling Technology, Boston, MA, USA) and β-actin (1:2000; 8457S,Cell Signaling Technology) and primary rabbit polyclonal antibodies against light chain (LC) 3-I/II (1:800; ABC929, Sigma), NQO1 (1:800; ab34173, Abcam, Cambridge, MA, USA),glutathione S-transferase (GST) (1:1000; ab19256, Abcam),and SOD1 (1:1000; ab20926, Abcam) were used. GAPDH and β-actin were used as internal references. After washing three times with Tris-buffered saline containing Tween 20, membranes were incubated with horseradish peroxidase-conjugated polyclonal secondary antibody of the appropriate species(1:2500; Santa Cruz Biotechnology) for 1 hour at room temperature. Protein detection was performed using an enhanced chemiluminescence detection kit (Jiancheng Bioengineering Institute, Nanjing, China). Gray value ratios of western blots were quantified by ImageJ software (NIH, Bethesda, MD,USA) and are expressed as the gray level ratio of the target protein relative to the internal reference protein.
Real-time quantitative polymerase chain reaction
Total RNA from cells was isolated using an RNeasy kit (Ta-KaRa Biotechnology, Dalian, China) following the manufacturer’s protocol. cDNA was prepared using a High Capacity cDNA Reverse Transcription kit (TaKaRa Biotechnology)according to the manufacturer’s protocol. Real-time quantitative polymerase chain reaction (qRT-PCR) analysis was performed using a Taqman primer and probe set from Applied Biosystems. The primer sequences are shown in Table 1. PCR reactions were run in triplicate in three independent experiments. β-Actin served as an internal control. Relative fold changes in mRNA expression were calculated using the formula, 2-ΔΔCt(Livak et al., 2001).
Immunocytochemistry
Cells were fixed in 4% formaldehyde, gently washed once with phosphate buffered saline, permeabilized in 0.1% Triton X-100 and then blocked in phosphate buffered saline with 1% bovine serum albumin for 1 hour. Cells were thenincubated with primary mouse monoclonal antibody against Nrf2 (1:100; Santa Cruz Biotechnology) for 2 hours at room temperature. After incubation with primary antibody, cells were incubated with a fluorescent goat anti-mouse polyclonal secondary antibody (1:200; Santa Cruz Biotechnology)for 30 minutes at room temperature. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). A fluorescence microscope (Leica, Wetzlar, Germany) with a 20 × objective was used to observe cells.

Table 1 Primers used for real-time quantitative polymerase chain reaction analysis
Total antioxidant capacity assay
Total antioxidants were measured using a total antioxidant capacity assay kit (Jiancheng Bioengineering Institute) in accordance with the manufacturer’s instructions. Briefly,cells were sonicated and centrifuged at 10,000 × g at 4°C.Supernatant aliquots were stored at -80°C for subsequent examination. Uric acid was prepared as a standard. Reactions were performed in a 96-well microtiter plate and absorbance was obtained by reading the plate at 520 nm.
Nitric oxide/nitric oxide synthase assay
Total nitric oxide (NO) and nitric oxide synthase (NOS) activities were measured using a NO/NOS Assay Kit (Jiancheng Bioengineering Institute) according to the manufacturer’s instructions. The NO assay was based on the ability of nitrate reductase to convert nitrate to nitrite. Kit reagents then convert nitrite to a colored compound that has a strong absorbance at 550 nm. Optical densities of reacted samples were measured at 550 nm. In the NOS assay, NOS catalyzes the production of NO from L-arginine. NO generated by NOS then undergoes a series of reactions and reacts with Griess reagent (Beyotime Biotechnology, Shanghai, China) to generate a colored product, which can be measured at 530 nm.
Keap1 siRNA transfection
Human Keap1 siRNA was synthesized and purified as previously described (Youn et al., 2015). Primer sequences were: Forward: 5′-GGC CUU UGG CAU GAA CTT-3′,Reverse: 5′-GUU CAU GAU GCC AAA GGC CTG-3′. Using solid-phase peptide synthesis, a myristic acid conjugated, cell-penetrating peptide (transportan) equipped with a transferrin receptor-targeting peptide (myr-TP-Tf) was prepared. The Keap1 siRNA oligonucleotides were annealed together and then mixed with myr-TP-Tf peptide at a 20:1(peptide to siRNA) molar ratio. SH-SY5Y cells at a density of 20,000 cells/mL were transfected with Keap1 siRNA-peptide complex for 3 hours and further incubated in complete culture medium. After siKeap1 transfection, cells were treated with 60 ng/mL TSA for 24 hours. Western blot analysis and qRT-PCR were used to assess the expression levels of Keap1 and Nrf2 in Control, siNC, TSA, siKeap1, and TSA +siKeap1 groups.
Statistical analysis
Data are expressed as the mean ± SD. Statistical analysis was performed using SPSS 17.0 software (SPSS, Chicago,IL, USA). Statistical evaluation was performed using oneway analysis of variance followed by Tukey’s post hoc test for multiple comparison. P < 0.05 was considered statistically significant.
Results
Protective effect of TSA on Aβ-induced cell toxicity is mediated via the Keap1-Nrf2 pathway
Cell viability of SH-SY5Y cells pre-treated with TSA at various concentrations was evaluated by MTT assays at various time points. The concentration of TSA (Figure 1A) and duration of incubation (Figure 1B) had little effect on SHSY5Y cell viability. Accordingly, three concentrations of TSA(low = 20, moderate = 40, and high = 60 ng/mL) were selected for the following experiments. Cells treated with Aβ25-35displayed low viability (Figure 1C) and abnormal morphology (Figure 1D), indicating that Aβ25-35strongly inhibited the growth of SH-SY5Y cells. However, groups pretreated with TSA all displayed better tolerance to the cell toxicity induced by Aβ and displayed near normal phenotypes (Figure 1D). There was a dose-dependent trend for increased cell viability with increasing TSA concentration (Figure 1C).Western blot assay (Figure 1E and F) and qRT-PCR (Figure 1G) results were consistent and suggested a potential role of TSA in regulating the Keap1-Nrf2 signaling pathway. In the TSA groups, Keap1 protein/mRNA levels were decreased and Nrf2 protein/mRNA levels were increased in a dose-dependent manner with high concentrations of TSA exhibiting greater Keap1 reduction and Nrf2 increase. Based on the above data, the optimal TSA concentration and incubation duration for subsequent experiments were high concentration (60 ng/mL) and 24 hours, respectively.
TSA increases nuclear Nrf2 levels and upregulates Nrf2 target genes, NAD(P)H quinone dehydrogenase 1 (NQO1),SOD1 and GST
Keap1 constitutively suppresses Nrf2 activity to a basal level.However, oxidative stress or chemical stimulation can affect Keap1 activity. This can, in turn, prevent Nrf2 degradation(Konstantinopoulos et al., 2011; Youn et al., 2014). Nrf2 accumulation leads to its translocation into the nucleus, which activates transcription of target cytoprotective genes that ensure cell survival (Dinkova-Kostova et al., 2002; Dinkova-Kostova and Talalay, 2008). To examine if Nrf2 levels in the cytoplasm and nucleus were altered by TSA treatment,we performed western blot assays on cytosolic and nuclear fractions. GAPDH and Lamin B antibodies were used as loading controls for cytosolic and nuclear fractions, respectively. Without TSA treatment, Nrf2 levels in the nuclear fractions were low in both control and Aβ-induced groups(Figure 2A). In groups pretreated with TSA, Nrf2 protein was clearly accumulated in the nuclear fraction and reduced in cytoplasmic fractions. Immunofluorescence staining to detect the location of Nrf2 showed consistent results (Figure 2D); greater co-staining of Nrf2 and DAPI was observed in cells pretreated with TSA compared with non-TSA-treated cells. These results indicated that TSA induced Nrf2 translocation into the nucleus.
To further determine whether TSA treatment changes Nrf2 target gene expression, protein/mRNA levels of representative genes, including NQO1, SOD1, and GST, were measured by western blotting and qRT-PCR, respectively(Figure 2B and C). TSA treatment noticeably increased NQO1, SOD1, and GST levels in both control and Aβ groups, indicating that TSA treatment disrupted Keap1-Nrf2 interaction, thereby releasing Nrf2 to translocate into the nucleus where it could activate/accelerate ARE-dependent detoxifying gene transcription.
Protective effect of TSA against oxidative stress
We next investigated whether TSA-mediated cytoprotection enhanced the antioxidant capacity of cells. Total antioxidant capacity was evaluated for control and TSA-pretreated SHSY5Y cells at 24 and 48 hours. As predicted, total antioxidant capacity was significantly enhanced in TSA-pretreated SHSY5Y cells compared with control cells (Figure 3A). The relationship between cell viability and H2O2concentration was then examined. Cell viability was attenuated in response to H2O2stimulation in a dose-dependent manner (Figure 3B).TSA-pretreated cells displayed higher viability compared with non-TSA treated cells at all H2O2concentrations. Cell viability of TSA-pretreated cells was increased 20% relative to control cells at 600 μM H2O2, and 14% at 200 μM H2O2.This observation indicated that the effect of TSA might be affected by the severity of oxidative stress. H2O2and Aβ treatment produced a substantial number of dead cells and abnormal cell morphology, while cells in TSA groups maintained relatively normal morphology (Figure 3C). The effects of treatments on NO content and NOS activity are summarized in Table 2. H2O2and Aβ dramatically increased NO/NOS levels compared with the control group, while TSA treatment reversed these changes, returning NO/NOS tobasal levels, especially in Aβ-induced cells.
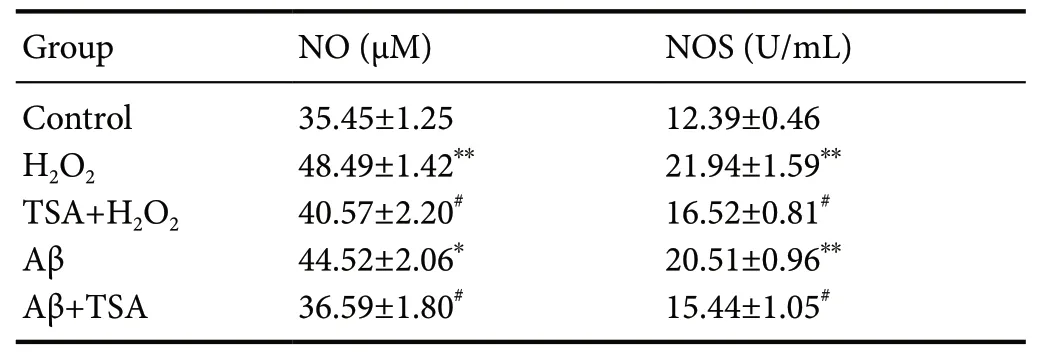
Table 2 Level of NO/NOS in SH-SY5Y cells
TSA alleviates Aβ-induced cell autophagy
Oxidative stress is a major inducer of autophagy, which is a self-degradation process that leads to cell cycle arrest.During autophagy, a cytosolic form of LC3 (LC3-I) is processed and conjugated to phosphatidylethanolamine to form LC3-II, which is recruited to autophagosomal membranes(Yue et al., 2009). Therefore, formation of LC3-II reflects autophagic activity. We, therefore, examined LC3-I and LC3-II levels by western blotting as a biochemical marker for autophagy. Six cell groups were prepared: control, TSA, Aβ,TSA + Aβ, H2O2, and TSA + H2O2. Increased LC3-II intensity was observed in both Aβ and H2O2groups compared with the control group, indicating increased autophagic activity(Figure 4). In the Aβ group, pretreatment with TSA resulted in significantly decreased LC3-II levels compared with no TSA treatment. Similar results were seen in the H2O2groups.These observations indicated that TSA plays an inhibitory role in autophagic activity induced by Aβ.
TSA reduces Keap1 protein levels at the post-transcriptional level
Western blot assays and qRT-PCR were performed to assess Keap1 levels in the following groups: control, siNC,TSA, siRNA, and TSA + siRNA. siNC is a negative control group for siRNA. Keap1 siRNA transfection reduced Keap1 mRNA levels by 50% and the addition of TSA decreased Keap1 mRNA levels further, by more than 75% (Figure 5A).Moreover, the reduction in Keap1 mRNA levels resulted in decreased Keap1 protein levels (Figure 5B). To confirm that Keap1 siRNA modulated Nrf2 expression, the mRNA and protein levels of Nrf2 were measured. As predicted, Keap1 knockdown resulted in upregulation of Nrf2 mRNA and protein levels (Figure 5). These findings indicated that TSA might activate Nrf2 through direct inhibition of Keap1.
Discussion
This study provides a novel explanation for the neuroprotective effect of TSA on AD. Currently, there is no effective way to prevent or cure AD; therefore, new therapeutic targets are urgently needed. The Nrf2 antioxidant pathway is an important cellular defense mechanism against oxidative stress and toxicity and may serve as a promising target for the treatment of neurodegenerative disease (Eades et al., 2011; Abed et al., 2015). Nrf2 activity is inhibited by two main negative regulators, Keap1 and glycogen synthase kinase-3 (Itoh et al., 1999; Rada et al., 2011). Recent findings indicate that the Keap1-Nrf2 pathway is a promising therapeutic target for AD and other neurodegenerative diseases (Abed et al., 2015;Kerr et al., 2017). Histone deacetylase inhibitors were recently suggested for AD treatment because they can improve learning and memory (Seo et al., 2013). In this study, TSA,a histone deacetylase inhibitor, inhibited Aβ-induced injury in SH-SY5Y neuroblastoma cells indicating that TSA may activate Nrf2 by inhibiting Keap1, thereby promoting Nrf2 target gene expression and activating a detoxification system.Amyloid plaques mainly consist of amyloid-β protein (Hardy and Selkoe, 2002). Targeting Aβ was predicted to be an effective strategy in AD therapy (Durairajan et al., 2012; Fan et al., 2015). AD patients have remarkably low gelsolin levels and gelsolin administration reduced amyloid load and decreased Aβ levels (Antequera et al., 2009; Yang et al., 2014a).Inhibitory effects of TSA can upregulate gelsolin levels (Yang et al., 2014b). TSA can also decrease cholesterol levels in neurons by modulating key genes involved in cholesterol synthesis (Nunes et al., 2013). Here, we explored whether TSA can modulate the Keap1-Nrf2 signaling pathway in Aβ-induced neurons. Instead of full-length Aβ peptide, a smaller 11-amino acid fragment, Aβ25-35, is often used experimentally as a convenient and mature alternative. Zhang et al. (2010) investigated the protective effects and the underlying mechanism of salidroside (Sald), an active compound isolated from a traditional Chinese medicinal plant, against Aβ25-35-induced oxidative stress in SH-SY5Y human neuroblastoma cells. Furthermore, Wang et al. (2010) investigated the underlying mechanism involved in the protective effect of astaxanthin, the most abundant flavonoid in propolis, on Aβ25-35-induced cytotoxicity in SH-SY5Y cells. In the present study, TSA treatment protected SH-SY5Y cells against reduced viability induced by Aβ. TSA also down-regulated Keap1 and up-regulated Nrf2. Finally, we examined whether the up-regulation of Nrf2 activated its target genes. Degradation of Keap1 results in Keap1-Nrf2 complex dissociation and Nrf2 translocation to the nucleus (Itoh et al., 1999; Konstantinopoulos et al., 2011). Nuclear Nrf2 binds to AREs on target gene promoters and activates gene transcription (Itoh et al., 1999). Hence, Nrf2 levels in the cytoplasm and in the nucleus were analyzed. Higher Nrf2 levels in the nucleus and correspondingly lower levels in the cytoplasm were observed in cells treated with TSA compared with cells not treated with TSA. These findings were supported by immunostaining, which showed co-localization of Nrf2 and the nuclear marker, DAPI. The levels of Nrf2 target genes were then examined, including NQO1, SOD1, and GST, which encode antioxidant enzymes. mRNA and protein levels of these genes were enhanced in TSA pretreated samples. These enzymes are involved in many antioxidant actions and catalyze a wide range of chemical detoxification (Dinkova-Kostova and Talalay, 2008). Together, these findings support the notion that a TSA-mediated effect on Keap1 causes Nrf2 accumulation, leading to Nrf2 translocation to the nucleus and consequently activation of its target genes.
TSA activates ARE-dependent genes, most of which areantioxidant enzymes; therefore, we examined the effect of TSA on total antioxidant capacity of cells after stimulation with H2O2. TSA increased total antioxidant capacity, indicating its role in defending against oxidative stress. H2O2decreased cell viability in a dose-dependent manner and TSA treatment inhibited this H2O2toxicity. Cells pretreated with TSA exhibited high cell viability and near normal cell morphology. Oxidative stress stimulates neuronal damage via the overexpression of NOS, which can increase the production of NO (Butterfield et al., 2007). A new bioactive molecule targeting neuronal nitric oxide synthase can attenuate the pathogenesis and progression of neuronal diseases (Maccallini and Amoroso, 2016). Effects of TSA on NO content and NOS activity were examined in this study.We established that TSA reversed the changes in NO/NOS induced by Aβ and H2O2. TSA enhancement of antioxidant capacity can be explained by Nrf2 activation, which induces ARE-dependent genes to maintain cellular redox homeostasis. Collectively, the results of this study demonstrate that TSA enhanced total antioxidant capacity and maintained NO/NOS at a normal level to protect cells against oxidative stresses, which are involved in the pathogenesis of almost all neurodegenerative diseases, including AD. Oxidative stress can induce autophagy, so we examined the effect of TSA on autophagy activity. Autophagy is a lysosomal degradation process that is essential for maintaining cellular homeostasis by clearing damaged organelles and waste proteins (Tan et al., 2014; Fan et al., 2015). Alterations in autophagy activity

Figure 1 Protective effects of TSA on 25 μM Aβ25-35-induced cytotoxicity in SH-SY5Y cells via regulation of Keap1 and Nrf2 expression.

Figure 2 TSA treatment triggers translocation of Nrf2 to the nucleus and simultaneously upregulates the expression of Nrf2 target genes.
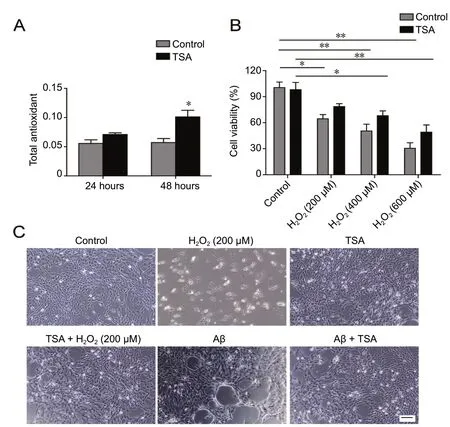
Figure 3 Protective effect of TSA (60 ng/mL) against oxidative stress in SH-SY5Y cells.
have been implicated in neurodegenerative disorders such as AD (Tan et al., 2014). Aβ can induce autophagy both in vitro and in vivo (Fan et al., 2015; Chellian et al., 2017). Autophagy was determined by detecting the biochemical markers,LC3-I and LC3-II. Consistent with previous studies, Aβ and H2O2 induced autophagy activity. Reduced LC3-II formation and high levels of LC3-I expression were observed in TSA groups, indicating that TSA can attenuate Aβ-induced or H2O2-stimulated autophagy activity in SH-SY5Y cells. In conclusion, the cytoprotective effect of TSA against Aβ is related to enhanced capacity against oxidative stress and inhibition of autophagy activity.
Genetic knockdown of Keap1 activates the Nrf2-ARE pathway (Jain et al., 2010). The microRNA, 200a, regulates Keap1 mRNA at the post-transcriptional level (Eades et al.,2011). Our results indicate that the TSA-induced reduction in Keap1 occurs at the post-transcriptional level because Keap1 mRNA levels were reduced further by TSA after Keap1 siRNA knockdown. Downregulation of Keap1 mRNA levels by TSA directly affects Nrf2 activity, further indicating that endogenous Keap1 levels are important for correct control of the Nrf2-ARE pathway.

Figure 4 TSA (60 ng/mL) treatment inhibits 25 μM Aβ23-25-induced autophagy activity.

Figure 5 TSA activates Nrf2 signaling by inducing Keap1 degradation.
We propose a novel mechanism in which TSA regulates the Keap1-Nrf2 pathway to protect cells against Aβ toxicity.TSA decreases Keap1 expression to activate the Nrf2-ARE pathway, which is an important antioxidant defense mechanism. TSA treatment attenuates cell damage induced by Aβ by enhancing antioxidant capacity and inhibiting autophagy activity. These outcomes open a new avenue of research regarding the potential neuroprotective role of TSA. However,the therapeutic effects of TSA should be further confirmed by in vivo animal experiments.
Author contributions:Study design: XRT, LHL, JJL; experimental implementation: LHL, WNP; data analysis: WNP, JJL; paper writing: YD,LHL. All authors approved the final version of the paper.
Conflicts of interest:The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support:None.
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
杂志排行

中国神经再生研究(英文版)的其它文章
- Ethanol extract from Gynostemma pentaphyllum ameliorates dopaminergic neuronal cell death in transgenic mice expressing mutant A53T human alpha-synuclein
- Peripheral nerve injury induced changes in the spinal cord and strategies to counteract/enhance the changes to promote nerve regeneration
- Genetic targeting of astrocytes to combat neurodegenerative disease
- Pathological significance of tRNA-derived small RNAs in neurological disorders
- Applications of advanced signal processing and machine learning in the neonatal hypoxic-ischemic electroencephalography
- Protective effect of hydrogen sulfide on oxidative stress-induced neurodegenerative diseases