含能晶体中分子间相互作用的特点及其启示
2020-09-17张朝阳
张朝阳
(中国工程物理研究院化工材料研究所,四川 绵阳 621999)
1 引言
分子间相互作用,从物理上讲,包括van de Waals作用力和静电作用力(库仑力);而化学上,它又被区分为氢键(HB)、卤键、π‑π 堆积、σ‑π、p‑π 等相互作用,这种区分主要是视具体的化学组成和结构而定的,常常无法严格描述其大小。如同一条条网线,分子间相互作用将含能分子或离子约束在固定的晶格里,它是形成含能晶体的原动力。因此,研究分子间的相互作用对认知含能晶体中分子或离子的堆积结构与这些堆积结构对含能材料(EM)性能的影响规律具有重要意义,这些认知将是EM 设计的理论基础。
目前,除传统的CHNO 分子外,聚合氮[1]、全氮离子[2-3]、含能离子盐[4-5]、含能金属框架(MOF)[6]、含能共晶[7]、含能钙钛矿[8]、高张力键能释放材料[9]及多种形式的复合体系[10]纷纷被制备出来,其中大部分体系常具有区别于传统CHNO 化合物的新组成与新结构。尽管如此,实际得到应用的EM 主流上仍然是由中性CHNO 分子构成的含能分子晶体,而分子间相互作用主要出现在这类晶体中。同时,目前大量出现了的含能离子盐,其大部分组成为有机离子,离子化程度远低于典型的无机离子,离子键作用并不突出,离子间的相互作用也可以用分子间相互作用的原理去很好地理解。
低感高能材料是EM 研究的一个重要方向,如何设计并合成低感高能化合物是获得这类材料的关键。显然,从已有含能化合物中认知分子间相互作用特点及其同宏观性能(如感度)间关系是获得相关设计原理或准则的基础。因此,本文将总结含能分子晶体与含能离子晶体(不包括无机离子盐体系)中分子间相互作用的特点,分子间相互作用对堆积结构、撞击感度和热安定性的影响规律,以及低(降)感高能晶体分子间相互作用对设计的启示,以期提供一个对含能晶体中分子间相互作用的较全面的认知,并有助于理解和设计低感高能化合物。
2 含能晶体中分子间相互作用的特点
目前尚无低感高能化合物的明确定义。先前我们考虑到TNT 和RDX 是现代EM 中里程碑式的代表,定义能量(爆速)高于RDX、撞击感度低于TNT 的化合物为低感高能化合物[11]。符合此定义的化合物非常有限,仅有如FOX‑7、NTO 和LLM‑105 及部分含能离子盐。鉴于此,本文以TNT 界定低感高能化合物,即,爆速高于TNT、撞感低于TNT 的化合物。这样,所涉及的体系将大大增加,有利于提供更多的物质结构信息。此外,有些化合物或晶体,经过共晶或成盐后,其撞感下降,我们称之为降感了的晶体。显然,这也是一种创制低感高能晶体的重要策略,有必要讨论之。当然,为了与低感高能晶体形成对比,讨论高感高能晶体中分子间相互作用也是必要的。
2.1 分子间氢键
含能分子和离子中常常存在H 原子,这为分子间HB 的存在奠定了元素条件。最近Ma 等系统研究了传统单质CHON EM 中的分子间氢键作用,包括11 种低感高能化合物TATB、NQ、DAAzF、DAAF、DATB、DNDP、NTO、TNA、α‑FOX‑7、LLM‑105 与TNB 和10种 敏 感EM ONDO、PETN、TNAZ、RDX、β‑HMX、BCHMX、ε‑CL‑20、BTF、HNB 和ONC[12-13],随 后,Bu等[14]又对EM 中的氢键作用进行了综述。分析表明,分子间HB 存在于所有11 种低感高能晶体中和大部分含H 高感高能晶体中,只有TNAZ 中不存在分子间HB,这也是导致其熔点较低的原因。如图1 所示,存在于低感高能晶体中的分子间HB 总体上强于高感晶体,表现出更短的HB 键长(d)、更高的平均键能(EHB)和总键能(ΣEHB)。低感高能晶体中更强的HB 可归结于其更强的HB 给体N─H,相对于高感晶体中的CH。一般而言,传统的由中性分子组成的含能晶体,其分子间HB 分布常常比较丰富(个数密度大),但按照Jeffrey对HB 强弱的分类原则[15],单个分子间HB 归属于弱氢键,即使在低感高能晶体中也是如此。
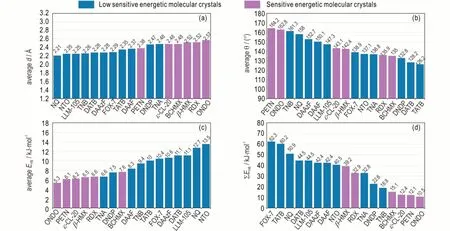
图1 传统的单组分含能分子晶体中分子间HB 的几何与能量Fig.1 Geometry and energy of intermolecular HBs in traditional single‑component energetic molecular crystals
相同结论也可以从含能分子共晶中得到。如图2所示,大部分CL‑20 基共晶中存在较丰富的分子间HB,当然,这些氢键单个上是比较弱的;同时,相比于ε‑CL‑20,大部分共晶中的分子间HB 增强(O…H 接触布居数增加),这表明分子间HB 的增强可能是这些共晶形成的驱动力[16]。与单组分的含能晶体相比,含能分子共晶中分子间相互作用类型及强度并未发生改变,HB 强度变化不大,因此,熵增加被认为是共晶形成的最主要的热力学因素[17]。此外,CL‑20基共晶中分子间HB的增强,也将是其撞感较CL‑20纯组分下降的原因,因为分子的解离发生在分子间相互作用消失之后,而增强分子间相互作用将在一定程度上迟缓分子分解。

图2 CL‑20 基共晶中以CL‑20 为目标对象的分子间原子间近接触布居数Fig.2 Populations of close intermolecular interatomic con‑tacts of the CL‑20 molecules in CL‑20‑based cocrystals
此外,Meng 等[18]还研究了含能离子盐中的分子间HB,重点关注了羟胺(HA)离子盐。如图3 所示,含能离子盐中分子间HB 一般显著强于传统含能分子晶体。含能离子盐中的阳离子,如HA+、NH4+、N2H5+、G+、AG+、DAG+和TAG+,通常为强HB 给体,它们中的大部分同时也是强HB 受体;此外,含能离子盐中分子间HB 属于典型的离子型HB,而含能分子晶体的HB为分子型的。这些原因导致含能离子盐中分子间HB强度大大提高,如图3 中更短的d,更大的EHB和ΣEHB。强分子间HB 将导致含能离子盐可能有高的堆积系数(PC)[18]和低撞击感度。

图3 含能离子盐与传统含能分子晶体中分子间HB 的比较Fig.3 Comparison in intermolecular HB between energetic ionic salts and traditional energetic molecular crystals
2.2 π⁃π 堆积
π‑π 堆积主要是针对含有π 键的分子或离子体系而言的,特别是含有大π 键的体系。由于组成EM 的许多分子为大π 键分子,这是π‑π 堆积的分子基础。由于大π 键分子中的电子离域效应,分子的稳定性增加,有助于相应EM 的低感度;因此,大π 键分子常常出现于低感含能晶体中。如图4 所示,大π 键分子中常含有H,但未必一定如此,例如无H 分子BTF、TASH和TAT 也有大π 键。

图4 含氢含能大π 键分子的正面与侧面视图Fig.4 Front and side views of planar π‑bonded structures of hydrogenous energetic molecules
π‑π 堆积模式可分为如图5所示的四种类型[12]。越来越多的例子表明,堆积模式对EM 的撞击感度可产生巨大的影响,这将在后面讨论。根据晶体中有无分子间HB的存在,可将之分为HB 协助的和无HB 的π‑π 堆积。图6 示出了一些典型含能晶体中HB 协助的面‑面π‑π 堆积,层内的分子间HB 支撑了这些层的稳定性;但未发现显著的层间HB 的存在。这些特点有利于滑移时保持层的完整性。因此,这些相互作用单元如HB 供体、HB 受体和它们间的作用方式必须在面‑面π‑π 堆积设计中认真考虑。如上所述,在传统CHON 的分子晶体中,分子间相互作用较弱,这种相互作用在设计中考虑起来比较困难;而在R1(R1=N 或O)─H…R2(R2=N 或O)相互作用中或含能离子盐中,分子间HB 将显著增强,将有助于晶体结构的理解与设计。一般的叠氮化物。TASH 和TAT 的分子堆积大大丰富了我们对无HB 协助的EM 晶体堆积的相关认知,但仍需要进一步的探索,例如,是什么支撑其面‑面π‑π 堆积的问题?是由于其大的分子结构?因为面‑面π‑π堆积的石墨的层结构就是一种分子尺寸大的极端情况;此外,尽管无分子间HB,但是由于叠氮基上的N原子所带电荷的类型存在不同,不同分子的叠氮基之间可存在一定的色散吸引与静电吸引作用,这可以是支撑其稳定层结构的物理基础。当然,这有待于进一步的确认。

图5 含能晶体中的四种π‑π 堆积模式及分子间/分子内势能(p)‑滑移距离(d)间关系,a 和b 分别表示滑移方向为左右与前后Fig.5 Four types of π‑π stacking in energetic crystals and inter/intra‑molecular potential(p)‑sliding distance(d)dependences of the four kinds of stacking. a and b denote the sliding along right/left and front/back,respectively

图6 含能晶体中HB 协助的面‑面π‑π 堆积. 每个子图中的上下分别表示层内的分子间HB(绿色虚线)和π‑π 堆积Fig.6 HB‑aided face‑to‑face π‑π stacking in energetic crys‑tals. The top and the bottom in each plot show the intralay‑ered intermolecular HBs represented by green dash and the π‑π stacking,respectively
2.3 分子间卤键
总体上,含卤素的EM 比较少见;但有时为了满足除剂的需要而将卤素引入EM。最近,三种含卤素的含能共晶被制备出来了,它们分别是DADP/TCTNB、DADP/TBTNB[22]和DADP/TITNB[23]。
其中,DADP/TITNB 是一个典型的通过共晶增强分子间相互作用与改善分子堆积模式而实现撞击感度较原有两种单组分都降低的例子。图8a 和图8b 分别示出了DADP/TCTNB 和DADP/TBTNB 中层内分子间相互作用为TCTNB 和TBTNB 相同分子间的卤键作用,即O…Cl 和O…Br,这与单组分的情形相似。相比于单组分,共晶中的卤键作用强度并无明显变化,因为DADP/TCTNB O…Cl 的R(卤键长与相关原子范德华半径之和的比值)在0.99~1.0 之间(图9a),与单组分TCTNB 的0.98~1.04 相差无几。而图8b 所示的O…Br的R在0.95~0.96 之 间,显 著 大 于TBTNB 单 组 分 的0.88~0.89,这表明共晶后,TBTNB 自身间的相互作用减弱,这也是DADP/TBTNB 不稳定的根本原因[22,24]。与前面两种共晶不同,DADP/TITNB 层内分子间相互作用包含了如图8c 所示的I…O 卤键(TITNB 的I 原子与DADP 中的O 原子)和C—H…O 的HB(TITNB 中的硝基O 原子与DADP 中的H 原子),特别是其I…O相对于单组分有大幅度的下降,从0.90~0.97 下降到0.84~0.94(图9c),表明TITNB 自身间的相互作用显著增强。同时,这也导致DADP/TITNB 的堆积模式显然不同于DADP/TCTNB 和DADP/TBTNB,表现更显著的分子间相互作用的各向异性:层状堆积中层内分子间相互作用显著强于层间相互作用(图8)。这表明增强分子间相互作用及其各向异性是导致其撞感降低的一个重要原因[23-24]。最近,Matzger 等[25]发现TITNB分子中I 原子存在一个最大的σ‑孔(σ‑hole),可以同许多其它的原子强烈作用而形成稳定体系。这可以作为一个重要的共晶策略。

图8 三种含卤共晶中层内(左)和层间(右)分子间相互作用,分别以绿色和紫色虚线示Fig.8 Intra‑(left)and inter‑(right)layered intermolecular in‑teractions of the three cocrystals represented by green andpurple dashes,respectively

图9 三种含卤共晶中卤键距离与其范德华半径之和的比值(R)Fig.9 Ratio(R)of the halogen bonding distances to com‑bined van der Waals radii of the three halogen cocrystals.
3 分子间相互作用对堆积结构与撞击感度的影响
含能晶体中分子间相互作用对其堆积结构产生显著的影响,即,分子相互作用增强将导致更紧密的堆积。例如,对于TNB 和其三种硝基衍生物TNA、DATB与TATB,它们的PC 随着越来越多的H 原子被氨基取代 而 增 加,分 别 是0.72、0.74、0.78 和0.79[26]。伴 随PC 和分子密度(dm)的增加,其堆积密度(dc)不断增加(dc=PC·dm),分 别 为1.676,1.773,1.801 g·cm-3和1.937 g·cm-3。密度的增加对提高EM 的性能是极为有利的。同时,由于分子结构与分子间相互作用的变化,这四种化合物的堆积模式变得越来越有利于剪切滑移,或者低撞击感度。在这四种具有类似分子结构的化合物中,分子内与分子间的HB 使得能量与安全性双重利好,使得EM 能量‑安全性矛盾大大缓解,这是我们要特别注意的[27-31],因为此种情形并不多见。
另外一个强分子间HB 有助于提高dc的例子是羟胺盐。Meng 等[18]通过比较分析发现,对于同一组具有相同阴离子的含能离子盐中,羟胺含能离子盐常常具有最高的dc和最高的PC(表1)。由于dc=PC∙dm,有必要明晰是PC,还是dm对dc的决定性作用。将一组具有相同BT2-含能离子盐的dm和dc列入表2 中,结果发现羟胺阳离子的dm显然低于G+、AG+、DAG+和TAG+,证实了羟胺盐的PC 对其高dc的决定性作用[18]。因此,如何通过增强分子间HB 来提高含能晶体的堆积密度以及其爆轰性能是低感高能化合物设计的一个重要内容。
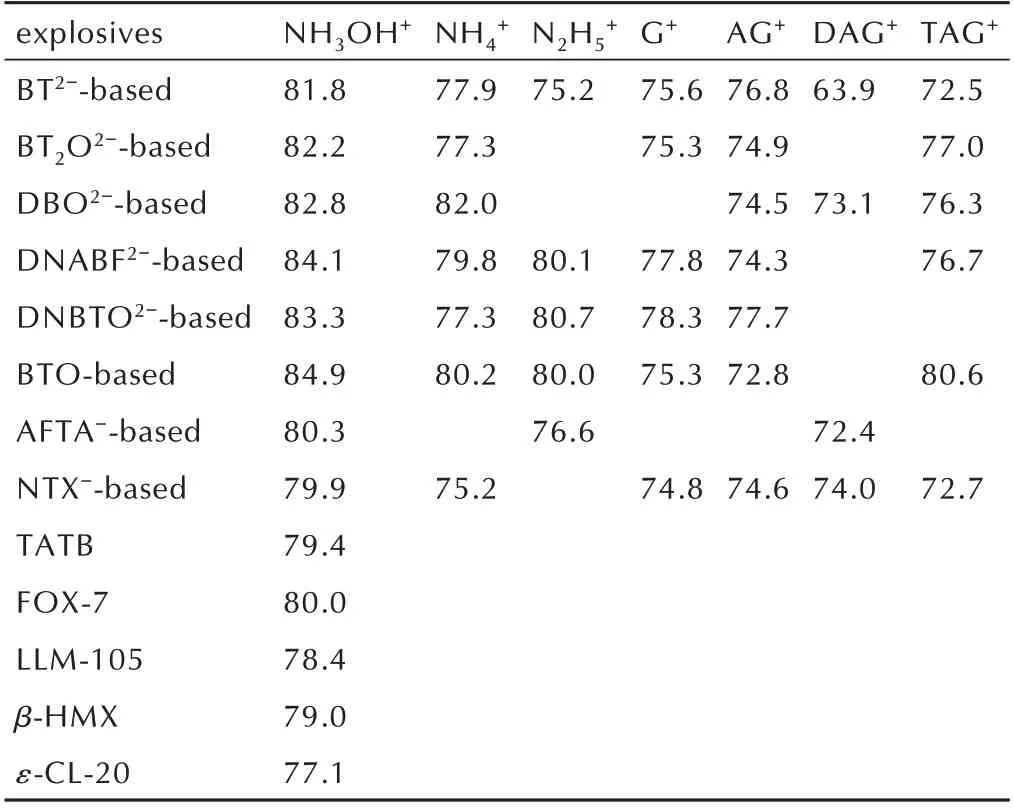
表1 含能离子盐与几种常见含能材料的PC(in%)的比较.Table 1 Comparison in PC(in%)among various energetic ionic salts and some common EMs.

表2 几种BT2-基含能离子盐的dc和其阳离子的dmTable 2 dm of various cations in BT2-‑based energetic ionic salts and their dc
分子间相互作用会影响含能晶体中分子堆积模式,进而影响其剪切滑移性能与撞击感度。事实上,具有π‑π 堆积结构的EM,其撞击感度较低,如图9 所示。最近几年,本课题组对π‑π 堆积模式对撞击感度的影响进行了深入研究,并提出通过改善分子堆积模式以缓解能量‑安全性矛盾,这也是含能晶体工程的重要内容[27-31]。Tian 等对π‑π 堆积模式对撞击感度影响的基本原理进行了阐述[31]。他们认为,感度机制是建立二者联系的桥梁。首先,冲击作用于EM 后,将产生压缩和剪切;此时,外界刺激将部分地转化为分子内与分子间增加的势能。随后,屈服与缺陷产生,缺陷处的温度显著升高,分子分解加剧,可形成热点并长大,实现最终点火。分子堆积模式将显著影响EM 的剪切滑移特性,从而影响热点的形成及后续点火:含能晶体越易滑移,能量越不易在晶体中积累,越有利于低感度[30]。
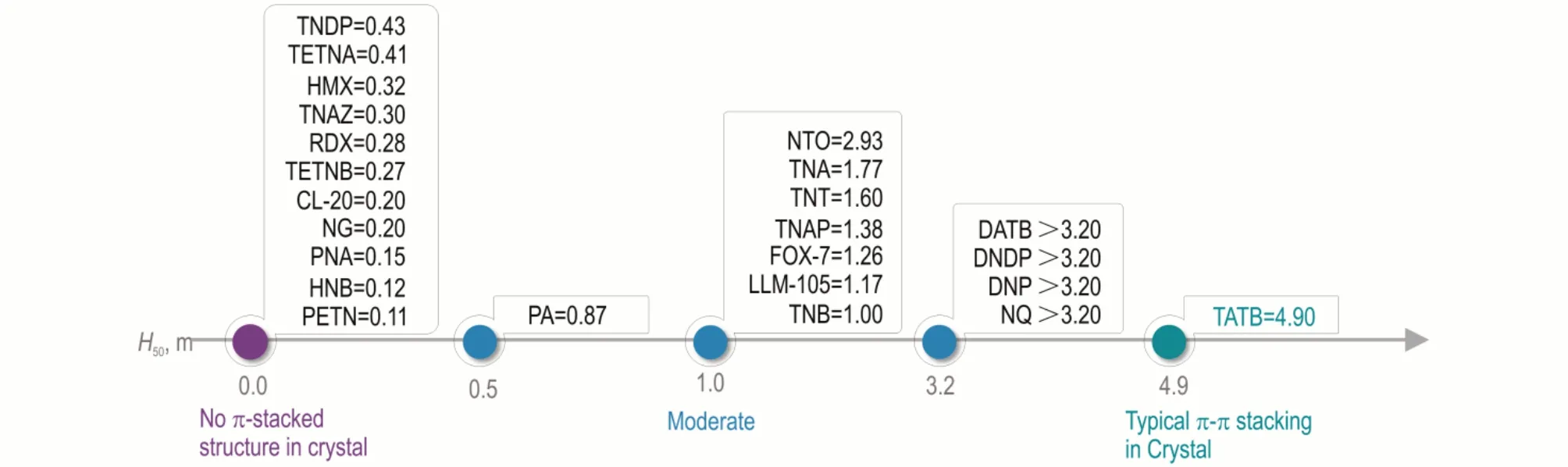
图9 π‑堆积结构与撞击感度的关系Fig.9 Relationship between π‑stacked structure and impact sensitivity of EMs
此外,相变导致的堆积模式的变化,将是决定EM表现出感度高低的决定因素之一。最近,Bu 等[32]研究了FOX‑7 低撞击感度的本质。从分子结构和堆积模式上看,FOX‑7 和LLM‑105 极具相似性,都是具有分子内HB 的大π 键分子和波形堆积。但是,LLM‑105表现出比FOX‑7 更高的耐热性(DSC 热解温度342 vs. 250 ℃)。因此,我们很难按已有理论理解FOX‑7和LLM‑105 具有相近的撞击感度。Bu 等认为,FOX‑7的力热导致相变及相关分子堆积模式改变,是其低感的重要原因之一。如图10 所示,无论是在加压还是升温的情况下,α‑FOX‑7 均可发生相变,且分子的堆积模式由波形堆积转变为面‑面堆积;而面‑面堆积模式是最有利于剪切滑移和低撞击感度的方式。因此,对于FOX‑7,相变使得其分子堆积更有利于低感;同时,这也表明对EM 受外界刺激时的结构变化的相关研究不容忽视,不能仅仅以常态下的结构来理解EM 的性能。

图10 FOX‑7 各晶型中的分子结构与堆积结构:(a)α‑(b)β‑(c)γ‑(d)α′‑ 和(e)ε‑型Fig.10 Molecular structures and crystal packing patterns of the polymorphs of FOX‑7:(a)α‑,(b)β‑,(c)γ‑,(d)α′‑ and(e)ε‑forms. The top,middle,and bottom in each shot show molecular structures,intralayered HBs represented by green dash and π‑π stacking,respectively
4 分子间相互作用对热安定性的影响
如前所述,增强分子间相互作用可提高晶体PC,也可以改善分子堆积模式,使其有利于剪切滑移和低撞击感度。但是,增加分子间相互作用并非一味地有利于改善EM 的性能,例如,分子间的强HB 可导致热安定性的恶化。这就是分子间HB 对EM 安全性影响的两面性[33]。最近,Lu 等发现了HA+和N2H5+基含能离子盐极低的热稳定性(如表3 所示的较低的热解温度)与强分子间HB 及H 原子易转移相关[33]。由于最小的体积和高活性,H 原子在EM 分解中扮演了重要角色[35-39]。

表3 系列含能离子盐的热解温度(Tdec,℃)的比较Table 3 Comparison in thermal decomposition temperatures(Tdec,℃)of series of energetic ionic salts
5 分子间相互作用对晶体设计的启示
从上可以看出,分子间相互作用的强弱与决定于分子间相互作用的分子堆积模式都是影响EM 能量和感度的重要因素。晶体设计大致包括两部分的含义:一是进行未合成分子设计,进而进行分子堆积得到晶体;一是通过对已知不同的分子/离子进行组合,获得共晶或盐。事实上,当前的晶体设计还存在极大的困难。例如,目前尚缺乏明确的分子结构与其堆积结构间的关系,遍历并获得一种分子的准确的晶体堆积结构需要海量的计算,一般的实验室难以承受。然而,He 等最近的研究表明,通过计算量较小的搜索能量占优的二聚体结构以确定可能的堆积模式也是可能的[40]。但从整体上来看,目前仍未有非常成功的面向低感高能晶体设计的例子,本节仅从已有的结构介绍相关的启示。
含能有机小分子可以分成两部分,一部分为中心的骨架部分,另一部分为外围的取代基部分。由于分子间相互作用主要通过不同分子取代基间的接触来实现,取代基在决定分子堆积模式中起到了重要作用。原则上,对于平面分子,π 分子骨架上的取代基可增加电子的离域程度,减少或增加骨架上的电子密度;进而影响整个分子的静电势及分子间相互作用。NH2是含能分子中一种代表性的取代基,引入NH2是匹配共轭体系中氧化性基团或吸电子基团(如NO2和N→O)并提高稳定性的重要策略。但需要指出的是,NH2的这种作用仅能在共轭体系中体现[11]。图11a‑d 示出了这种趋势,即随着对TNB 分子中H逐步被NH2取代,分子的平面度不断提高,分子在晶体中的堆积模式由混合型变成了面‑面堆积型,同时,分子的堆积系数与堆积密度不断提高。这表明,对于给定的分子骨架,同一种取代基的数量可影响分子的堆积结构。
同样,取代基的不同类型也可以影响分子在晶体中 的 堆 积 模 式,如 图11e~图11h 所 示:NTAZ 上 的H 原子为氨基、硝胺基和叠氮基取代后,分别形成了ANTA、DTAN 和ANTZ,其堆积模式由混合型堆积变化为交叉和面‑面堆积[41-44]。显然,不同取代基将导致分子稳定性与分子堆积模式产生差异,这也将导致其热稳定性与机械感度的差异。但是,这种取代基与分子稳定性及分子堆积模式间的关系研究仍然比较缺乏,最主要的问题是实验数据的缺乏。一旦高通量的晶体结构预测成为可能,这种分子结构与堆积结构间的关系将被建立起来,有助于我们准确进行晶体设计。

图11 取代基对晶体堆积模式的影响Fig.11 Substituent Effect on the crystal stacking type
通过共晶,含能材料的性质有望通过改变结构与组成而获得调节,因此也被认为是获得新型EM 的一种途径。例如,ANTA 本身是一种交叉型堆积的晶体,但同水共晶后,则为面‑面堆积,有利于剪切滑移及低撞击感度[41]。BTO/H2O[45]和DNPP/H2O[46]也有类似的面‑面堆积结构,撞击感度较单质组分有显著的降低。实际应用中,为避免过多的能量损失,常选择的共晶配体均为含能组分,此谓之为含能‑含能共晶。通过含能‑含能共晶,也可以实现通过改善分子堆积模式来降低撞击感度。如图12 所示,DNBT/ANTA 就是一种面‑面堆积的晶体,而它们的单组分分别是波形和交叉型的堆积[47]。这种面‑面堆积模式主要决定于平面的分子结构与浓密的分子间HB。
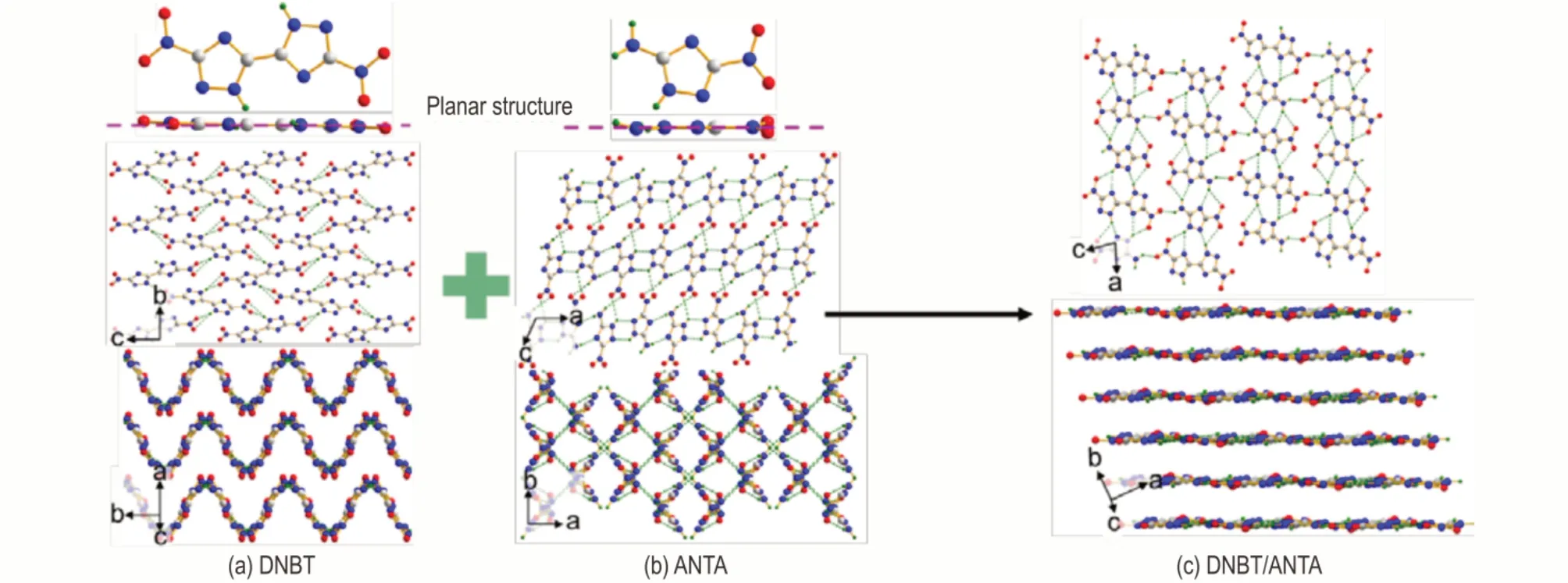
图12 通过共晶获得的π‑π 堆积结构Fig.12 Modification to face‑to‑face π‑π stacking by cocrystallization
成盐也是一种改善含能晶体堆积结构与性能的有效途径。同中性的含能分子相比,含能离子盐中常因富含C─N 键而具有较正的生成焓和较大的放热量;同时,离子化可增加分子稳定性、增强分子间相互作用与堆积密度。对于高能低感的含能离子盐的设计,Shreeve 课题组[30]曾按照我们提出的堆积模式‑撞击感度间的关系,设计并合成了低感的离子盐AFTA‑HA[48]。 同 样,如 图13 所 示,含 能 离 子 盐NNN‑G 和NNN‑AG 分别具有面‑面和波形的堆积[49]。NNN‑和G+都具有平面的分子结构,构成了NNN‑G面‑面堆积的基本条件;而AG+是非平面的,形成的波形堆积有平面堆积的趋势。这表明,相对于波形堆积的NNN,成盐既可改变也可保持这种堆积。

图13 Modification of π‑π stacking by salificationFig.13 通过成盐改变π‑π 堆积
6 结论
分子间相互作用是晶体中分子堆积的原动力,是决定EM 能量、安全性、力学性能等的本质因素,也是我们从低感高能化合物的本征结构——分子结构与晶体结构认知其组成、结构与性能及其相互关系的出发点。认知含能晶体中的分子间相互作用将有助于丰富含能化合物设计理论、提高低感高能化合物的设计效率和加快低感高能材料的创制。总之,含能晶体中分子间相互作用具有以下特点:(1)低感高能晶体一般具有强于高感高能晶体的HB;(2)面‑面π‑π 堆积是成就低感最具优势的分子堆积模式;(3)通过增强分子间相互作用并增加分子堆积的各向异性,是从晶体本征结构上实现降感的重要途径,更具有晶体工程的意义;(4)尽管增强分子间HB 有利于低撞击感度,但有可能导致热安定性下降,因为强HB为H转移准备了条件。
此外,分子间弱相互作用的准确描述、分子结构与堆积结构的关系及热力作用分子间相互作用的演化规律还有待于我们进一步研究,因为这些内容是我们探索EM 组成、结构与性能关系和设计新材料的基础。

附表 文中化合物名称及代号

续表
