Aun(n=2-7)在金红石型TiO2(110)面吸附的理论研究*
2020-09-17夏树伟杨继宏于良民
夏树伟,杨继宏,于良民
(中国海洋大学 海洋化学理论与工程技术教育部重点实验室,化学化工学院,山东 青岛 266100)
二氧化钛(TiO2)是综合性能优良半导体材料,被广泛应用于光催化、太阳能电池、涂料、净化处理等领域。20世纪70年代藤嶋等发现光照下金红石型二氧化钛能使水持续产生氢气[1],吸引众多科研工作者在TiO2光催化领域进行了深入研究。TiO2在自然界中以金红石、锐钛矿和板钛矿形式存在,禁带宽度分别为3.0,3.2,3.1 eV,其中金红石晶型最稳定[2-3]。Labat等人计算发现金红石相TiO2(100),(001)面的能量高于(110)面,(110)面最稳定[4]。二氧化钛只能响应紫外光(约占太阳光总能量4%),极大地限制了其应用[5]。为了弥补这一缺点,研究集中在设法降低体系禁带宽度,常用的方法有离子掺杂、染料敏化、贵金属沉积及半导体耦合等[6],其中表面负载贵金属纳米粒子在光照下能够有效发生水光解反应,受到了极大关注。在光照射下纳米金团簇表面等离子体共振产生热电子,能够提高载流子密度和体系的光吸收能力[7]。本论文对金团簇在TiO2(110)面的吸附进行理论研究,探索了纳米金团簇对TiO2能带的影响机理。
1 计算模型和方法
建模与计算均由Materials studio软件完成。由Visualizer构建模型,几何优化及相应性质分析由CASTEP完成,前线轨道和静电势计算采用DMol3模块。CASTEP计算采用密度泛函理论DFT[8-9],GGA-PBE交换关联泛函,超软赝势。采用BFGS算法优化晶胞中原子坐标。TiO2晶胞优化布里渊区积分采用Monkhorst-Pack方法,K点3×3×5,截断能340eV。金红石型TiO2(110)晶面及吸附体系结构优化,K点2×1×1。采用DFT+U方法修正GGA-PBE计算半导体材料禁带宽度偏低的问题,取U=7.3 eV,能带间隙为2.54 eV。
体相金红石型二氧化钛晶胞参数取自实验值[10](a=4.594 Å,b=4.594 Å,c=2.959 Å,α=β=γ=90°),使用三维周期性边界条件,优化得到晶胞参数a=4.654 Å,b=4.654 Å,c=2.971 Å,α=β=γ=90°,与实测值误差小于2%。将金团簇Aun置于边长12 Å的立方体箱中优化。搭建了4×2×2的二氧化钛超晶胞,表面板模型一共6层,固定底部2层,真空层为15 Å。
2 结果讨论
2.1 金团簇结构
优化得到的Aun(n=2-7)金团簇的稳定结构和键长如图1(a)所示,可见稳定构型表现出一定的对称性。Au2中Au-Au键长2.56 Å,Au3中键长变长。Au4为菱形,四边键长2.71 Å略长于中心键长2.66 Å。Au5为梯形,内部键长2.80 Å略长于外部键长2.70 Å,2.65 Å。Au6为三角形,内部键略长。Au7为近似正六边形,内部与外部键长较接近。随着团簇的增大,平均键长逐渐增加(Au2的2.56 Å到Au7的2.75 Å)。以上结构与文献中其他方法得到的结果相吻合[10-11]。团簇表面静电势如图1(b)所示,红色代表正值,蓝色代表负值,表面静电势最大值分布于团簇顶点原子外侧的红色区域。
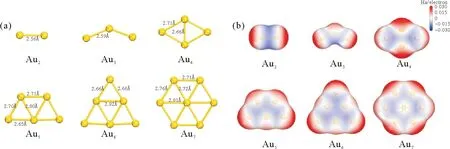
图1 Aun的(a)平衡几何构型和(b)表面静电势Fig.1 (a)Equilibrium geometry and (b)electrostatic surface potential of Aun
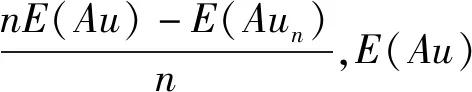
总能量二阶差分也可表征团簇稳定性,计算式为:Δ2E(n)=E(Aun+1)+E(Aun-1)-2E(Aun),由图2(b)看出,Δ2E随金原子数增加出现波动,金团簇稳定性呈奇偶振荡现象,即偶数原子团簇稳定性高于相邻奇数原子团簇,Au2和Au6稳定性较高。奇数原子团簇因存在未配对6 s电子,活性较相邻电子全配对的偶数原子团簇高,稳定性降低。
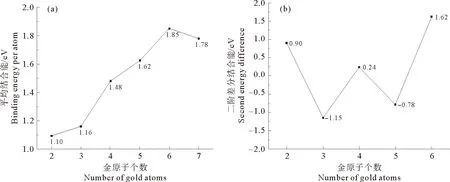
图2 Aun团簇的(a)平均结合能和(b)二阶差分能量Fig.2 (a)Bindingenergyper atom and (b)second energy difference of Aun clusters
由金团簇HOMO(最高占据轨道)和LUMO(最低空轨道)分布(见图3(a))可分析团簇活性位点。金团簇HOMO和LUMO轨道主要分布于团簇外围,表明外侧金原子易与TiO2表面发生相互作用进行吸附。HOMO/LUMO能级差(见图3(b))与分子的化学活性相关,能级差越小,活性越高。能级差也出现奇偶振荡现象:奇数原子团簇能级差小于偶数原子团簇能级差。可见能级差与能量二阶差分的结果一致,可以预测团簇的稳定性。
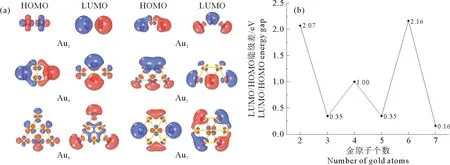
图3 Aun团簇的HOMO/LUMO(a)轨道分布和(b)能级差Fig.3 HOMO/LUMO(a)orbital and (b)energy gap of Aun clusters
2.2 吸附构型和吸附能
TiO2(110)面如图4所示,(110)面上有二配位氧原子O2c,三配位氧原子O3c,五配位钛原子Ti5c和六配位钛原子Ti6c。由于表面原子含悬空键,发生表面弛豫降低表面能。在弛豫作用下,表面原子在z轴方向发生了轻微的位移,Ti5c位移-0.25 Å,Ti6c位移0.30 Å,O2c和O3c仅位移约0.1 Å。表面静电势分析可知Ti原子静电势为正值,O原子为负值,其中表面O2c上负电荷密度最大,结合Aun团簇表面静电势分布可以预测Aun团簇应该通过团簇端位吸附在O2c位。
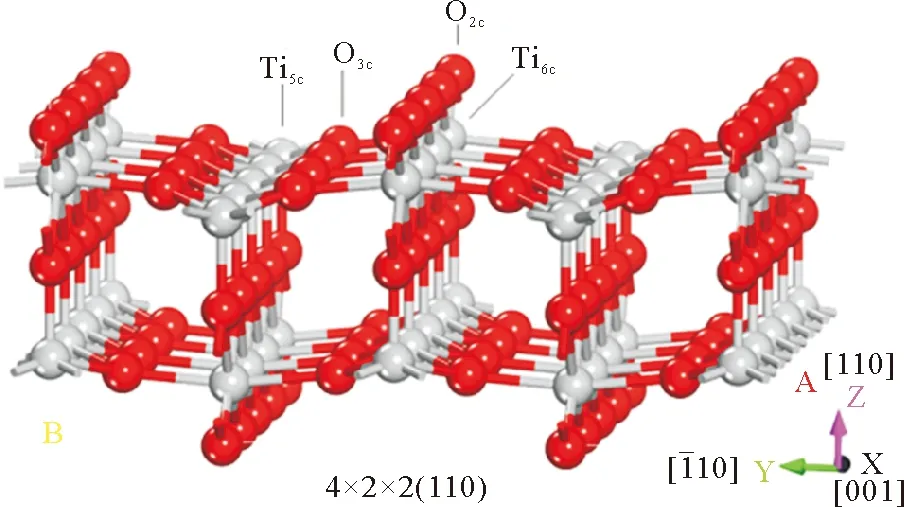
图4 TiO2(110)面的板模型Fig.4 Slab model of TiO2 (110) surface
通过研究单个金原子与TiO2(110)面的相互作用确定了表面活性吸附位点。TiO2(110)面上存在3种吸附位点,分别为顶位(T),桥位(B)和空位(H)。经优化得到稳定吸附构型为3种顶位吸附T(Ti6c),T(O2c)和 T(O3c)和2种空位吸附H(O2cTi6cO3cTi5c)和 H(O3cTi5c)。
金团簇在二氧化钛表面吸附能表达式:Ebind=E(Aun/TiO2)-E(TiO2)-E(Aun),E(AUn/TiO2)为稳定吸附体系能量,E(TiO2)为纯净TiO2能量,E(Aun)为游离金团簇能量。
由吸附能(见表1)可知T(O2c)是TiO2(110)面最稳定吸附位点,O2c位于(110)面的最外侧,易与Au原子形成化学键,这与文献[13-17]结果吻合。

表1 单个金原子稳定吸附构型的吸附能Table 1 Binding energy of single Au atom stable adsorption configurations
金团簇Aun在TiO2(110)面上各稳定吸附构型及其吸附能如图5所示。
金团簇在二氧化钛表面的吸附稳定性出现奇偶振荡现象,即偶数原子团簇吸附能较小,绝对值小于1.0 eV,奇数原子团簇吸附能较大,绝对值大于1.5 eV,因此奇数金原子团簇在二氧化钛表面的吸附作用较强,这与奇数原子团簇体系的活性较高有关。
由电荷密度分析可知金原子和氧原子之间的电荷密度0.05e/Å3,说明形成了化学键,因此金团簇通过键合O2c原子化学吸附于TiO2表面,吸附后金团簇的结构基本不变,仍表现为高对称性(见图5)。吸附稳定性最优的是Au3,其金原子距离表面氧原子约为2.15 Å,吸附能为-2.09 eV。在Au3/TiO2,Au5/TiO2和Au7/TiO2吸附体系中,吸附所形成的Au-O键长均小于2.2 Å,而Au2/TiO2,Au4/TiO2和Au6/TiO2体系中Au-O键长则大于2.3 Å。已知键长越短键能越高,因此奇数原子金团簇吸附较偶数原子金团簇吸附更为稳定,这与吸附能的结果一致。
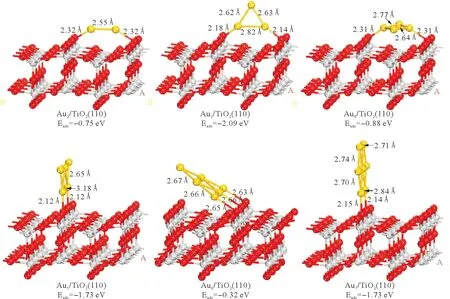
图5 Aun团簇在TiO2(110)面上的稳定吸附构型和吸附能Fig.5 Stableadsorption configurations and adsorption energy of Aun on TiO2(110) surface
2.3 吸附前后的能带、态密度
采用GGA+U方法计算了体相TiO2、TiO2(110)面和Aun/TiO2(110) 吸附体系的能带结构(见图6),当Ti原子3d轨道U=7.3 eV时,体相金红石型TiO2禁带宽度Eg=2.54 eV,纯净TiO2(110)面禁带宽度较体相TiO2略有增大,为3.17 eV。吸附金团簇后体系能带结构明显改变,奇数金原子体系中因存在单电子,能带发生分裂。Au2/TiO2体系禁带宽度1.88 eV,在纯净TiO2禁带中引入了新能级,即能带结构中的水平线段,导带底降低至2eV左右。Au3/TiO2体系禁带宽度1.92 eV,导带底在小于2.0 eV处出现新能级。Au4/TiO2体系禁带宽度降为1.47 eV,TiO2价带顶之上出现新能级,导带底下移至0.8 eV。Au5/TiO2体系中能带间隙约1.89 eV,与Au2/TiO2、Au3/TiO2体系的能隙相近。Au6/TiO2的能带结构与Au4/TiO2相似,带隙为1.43 eV。Au7/TiO2带隙为1.35 eV,导带底出现新能级。可见吸附金团簇后TiO2禁带宽度明显降低,使其吸收光由紫外区进入可见区,大大提高了对日光的利用率。
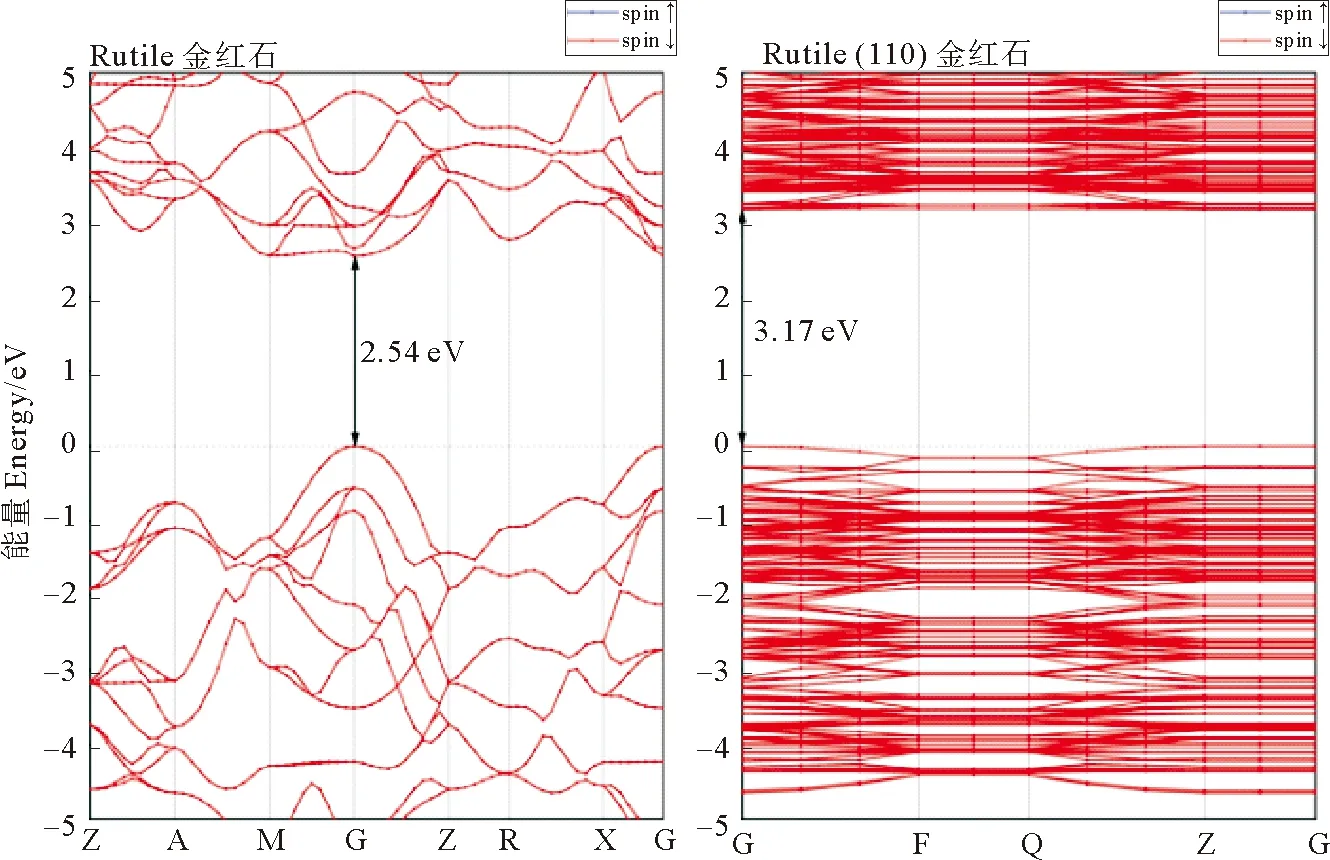
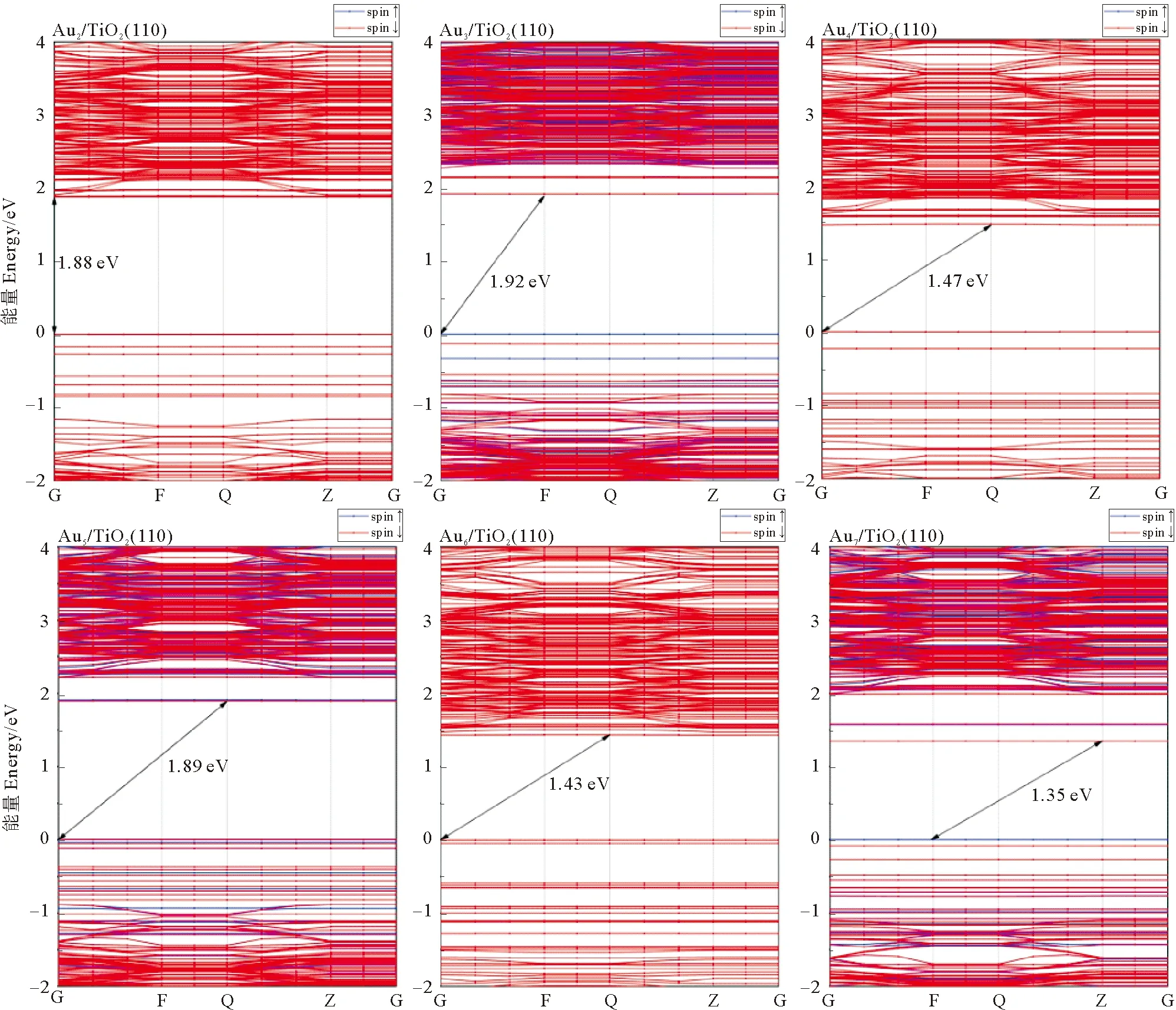
图6 体相TiO2、TiO2 (110)面和吸附Aun的TiO2 (110)面的能带结构Fig.6 Band structure of TiO2,TiO2(110) surface and Aun adsorption on TiO2(110) surface
3 结论
基于密度泛函理论,研究了金团簇Aun(n=2-7)在TiO2(110)面的吸附及其对TiO2能带结构的影响。结果表明,表面O2c原子为活性吸附位点,优化获得Aun(n=2-7)在TiO2(110)面的稳定吸附构型,偶数原子金团簇近似平行于TiO2(110)面,而奇数原子金团簇则近似垂直于表面。Au原子与TiO2表面形成的Au-O2c键是决定吸附稳定性关键因素。能带分析表明,金团簇在TiO2的禁带中引入新能级,有效降低禁带宽度,提高光催化性能。
