含有吡啶基的油溶性稠油降黏剂合成与性能评价*
2020-09-10王秋霞王弘宇周法元王曼依宁一凡
王秋霞,王弘宇,张 华,周法元,刘 昊,王曼依,宁一凡
[1.中海石油(中国)有限公司天津分公司,天津 300459;2.中国石油大学(华东)石油工程学院,山东 青岛 266580]
中国的稠油资源极为丰富,已在12个盆地发现了70多个重质油田,预计储量超过3×1012t[1]。在当前国内原油对外依存度不断攀升的情况下,稠油的高效开发对于保障国家能源安全、推进社会经济发展具有重要的意义[2]。提高稠油开发采收率的关键之一在于降低稠油的黏度,这是因为,稠油黏度的降低可以改善水油流度比,避免注水开发过程中,注入水突破过早,从而扩大注入水的波及范围[3-5]。化学降黏是指向稠油中加入化学药剂而降低稠油黏度,使用的药剂包括水溶性降黏剂和油溶性降黏剂。与水溶性降黏剂相比,油溶性降黏剂具有耐温性好、适用范围广、对采出液处理影响小等优点[6-8]。
胶质、沥青质是含有杂原子的多环芳核或环烷芳核,侧链上带有羟基、胺基、羧基、羰基等极性基团结构复杂的大分子化合物。稠油中的胶质、沥青质、金属化合物通过氢键、π-π作用、配位作用等产生很强的相互作用,形成类似于晶体结构的缔合体、超分子胶团等结构,使得稠油具有很高的黏度[9-11]。由此可见,油溶性稠油降黏剂需要能够很好地降低胶质、沥青质之间的相互作用,拆散聚集体结构,才能达到良好地降低稠油黏度、改善稠油流动性的效果。
作者以4-乙烯基吡啶(VP)、苯乙烯(ST)、马来酸酐(MA)、丙烯酸十八酯(SA)共聚,制备了一种新型的稠油降黏剂,并对其降黏性能进行了评价。在常规油溶性降黏剂分子结构的基础上引入吡啶基团,一方面利用吡啶的N原子和芳香性性质增强药剂拆散胶质、沥青质之间的氢键和π-π作用的能力;另一方面,稠油中存在少量的高价金属离子,这些离子可以和稠油中带有孤对电子的原子(比如N、O等)通过配位作用形成空间网架结构,因此,利用吡啶的孤对电子与金属离子之间可以形成配位结构的性质,提升药剂拆散稠油中有机质与高价金属离子之间配位作用的能力。为稠油油藏化学降黏体系的研发提供参考。
1 实验部分
1.1 原料与试剂
实验用稠油:50 ℃下黏度为1 850 mPa·s,现场用油溶性降黏剂(HYR)、煤油:中海油天津分公司提供。
VP、ST、MA、SA、过氧化苯甲酰(BPO)、甲苯:上海阿拉丁生化科技股份有限公司。
1.2 降黏剂合成
在装有冷凝器、温度计、搅拌器的三口烧瓶中加入一定量的VP、ST、MA、SA和甲苯,升温至50 ℃,搅拌待所有单体溶解后,加入BPO,通入氮气除氧30 min后,将温度升高进行聚合反应。待反应结束后,将产物转移至烧杯中,冷却至室温,用甲醇洗涤3次,将产物真空干燥,得到降黏剂。
1.3 降黏剂表征
使用德国Bruker公司TENSOR 27型傅立叶转换红外光谱仪(FTIR)定性分析样品的分子组成和结构,样品经溴化钾压片后,选择收集频率为400~4 000 cm-1。
1.4 降黏剂性能评价
使用NDJ-8型数字显示旋转黏度计进行测量,实验步骤为(1)将降黏剂溶于煤油中,w(降黏剂)=1%;(2)用水浴将原油加热至50 ℃;(3)将降黏剂煤油溶液加入至原油中[m(煤油)∶m(稠油)=1∶9],搅拌50 min;(4)分别测定原油加入降黏剂前后的黏度。降黏率按照式(1)计算。
(1)
式中:E为降黏率,%;μ0为原油初始黏度,mPa·s;μr为加降黏剂后的原油黏度,mPa·s。
2 合成参数优化
2.1 单体用量优选
在降黏剂的合成过程中,选取VP、ST、MA、SA作为单体。其中,SA和ST为亲油性单体,VP和MA具有一定的亲水性,因此,为了保证产品具有良好的油溶性,将SA和ST作为主要原料;ST的苯环可以拆散胶质、沥青质之间的π-π作用;MA的氧原子可以拆散胶质、沥青质之间的氢键作用;VP的吡啶环可以拆散胶质、沥青质之间的π-π作用和氢键作用[12-13]。由此可见,对于降黏剂而言,单体配比对其性能具有重要的影响,这是因为单体配比的变化,一方面影响到产品的溶解性能,另一方面还影响到产品拆散胶质、沥青质相互作用的能力。
通过正交实验考察了4种单体的加量对于产物降黏性能的影响,结果见表1。实验中m(单体)∶m(甲苯)=2∶8,m(BPO)∶m(单体)=0.2%,反应温度为70 ℃,反应时间为6 h。
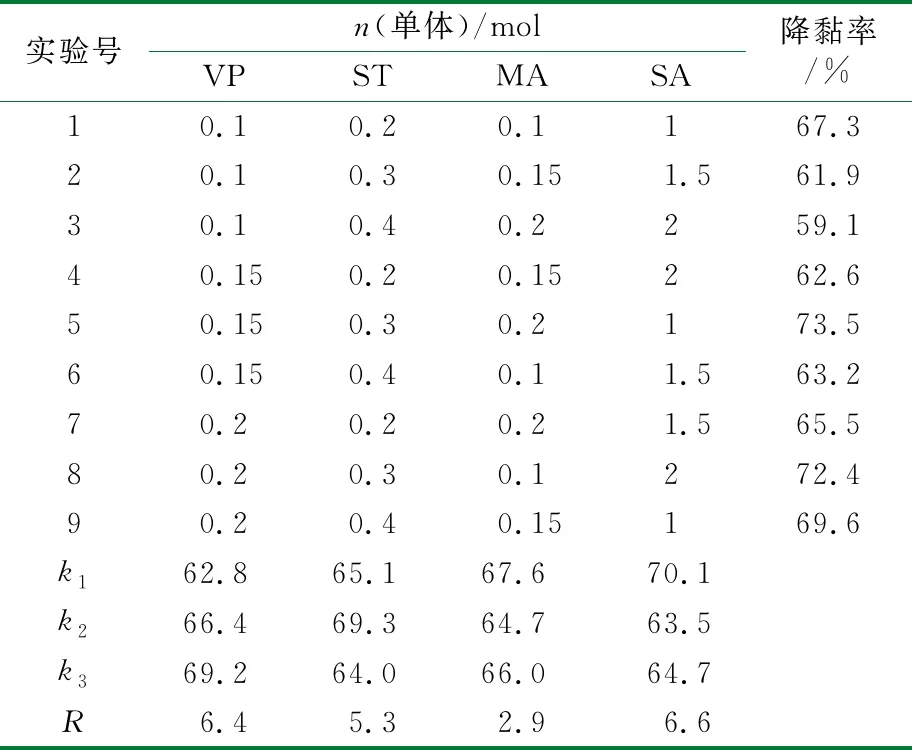
表1 正交实验样品性能评价结果
通过正交实验结果中各水平各因素对产物降黏性能影响的加权平均值(k)及极差(R)分析可知,n(SA)和n(VP)对于产物降黏性能的影响接近,高于n(ST)和n(MA)的影响。每种单体取最优的加权平均值,得到最佳n(VP)∶n(ST)∶n(MA)∶n(SA)=0.2∶0.3∶0.1∶1。
VP作为新单元引入常规降黏剂中,其对于产物性能影响较大,因此,在正交实验优选的前提下,n(ST)=0.3 mol,n(MA)=0.1 mol,n(SA)=1 mol,m(单体)∶m(甲苯)=2∶8,m(BPO)∶m(单体)=0.2%,反应温度为70 ℃,反应时间为6 h,进一步考察n(VP)对产物降黏性能和反应产率的影响,见图1。
由图1可知,当4种单体分别为正交实验所得最佳用量时,产物的性能优于正交实验合成各组样品的结果。当n(VP)增加时,产物的降黏性能呈现先增加再减小的趋势,n(VP)=0.25 mol,产物的降黏性能最佳。n(VP)=0.15~0.25 mol,反应的产率较高,当n(VP)=0.3 mol,产率显著下降。当n(VP)>0.25 mol,产物的产率降低,这说明VP的聚合活性较低,用量过大时,单体的转化率降低或者体系中形成大量的低聚物,反应产率降低。当n(VP)>0.25 mol,产物的降黏性能降低,这可能是由于VP的极性较大,用量过大时,产物的亲油性较弱,降低了聚合物与原油的亲和能力,从而使得降黏率降低。因此,结合产物降黏率和反应产率,优选n(VP)=0.25 mol。

n(VP)/mol图1 n(VP)对产物降黏率和反应产率的影响
2.2 甲苯用量优选
n(VP)=0.25 mol,n(ST)=0.3 mol,n(SMA)=0.1 mol,n(SA)=1 mol,m(BPO)∶m(单体)=0.2%,反应温度为70 ℃,反应时间为6 h,考察m(单体)∶m(甲苯)对产物降黏性能和反应产率的影响,见图2。

m(单体)∶m(甲苯)图2 m(单体)∶m(甲苯)对产物降黏率和反应产率的影响
由图2可知,当m(单体)∶m(甲苯)从1∶9增加至3∶7时,产物降黏率和反应产率持续上升,当m(单体)∶m(甲苯)进一步增加,降黏率出现下降。在自由基聚合中,增加m(单体),可以提升聚合反应的速率、提升产物的分子量,从而对产物性能和反应产率产生影响;然而,自由基聚合是放热反应,单体浓度越高,反应中后期体系的温度越高,温度升高对于聚合物分子量的增加是不利的,此外,温度过高还会导致聚合过程剧烈,甚至出现爆聚的现象[14]。因此,结合产物的降黏率和反应的产率,同时考虑反应的平稳程度,优选m(单体)∶m(甲苯)=3∶7。
2.3 引发剂用量优选
n(VP)=0.25 mol,n(ST)=0.3 mol,n(MA)=0.1 mol,n(SA)=1 mol,m(单体)∶m(甲苯)=3∶7,反应温度为70 ℃,反应时间为6 h,考察m(BPO)∶m(单体)对产物降黏性能和反应产率的影响,见图3。

m(BPO)∶m(单体)/%图3 m(BPO)∶m(单体)对产物降黏率和反应产率的影响
由图3可知,随着m(BPO)的增加,反应的产率持续增加,而产物的降黏率先增加后下降。提升引发剂的加量,引发剂分解生成的初级自由基增多,一方面单体被引发聚合生成大分子的可能性增加,反应产率提高;另一方面,聚合反应速率提高,反应温度升高,产物分子量降低,这可以影响到降黏剂的降黏性能[15]。因此,优选m(BPO)∶m(单体)=0.3%。
2.4 反应温度优选
n(VP)=0.25 mol,n(ST)=0.3 mol,n(MA)=0.1 mol,n(SA)=1 mol,m(单体)∶m(甲苯)=3∶7,m(BPO)∶m(单体)=0.3%,反应时间为6 h,考察反应温度对产物降黏性能和反应产率的影响,见图4。

t/℃图4 反应温度对产物降黏率和反应产率的影响
BPO需要在一定的温度下分解,才能提供合适的初级自由基浓度,从而保证聚合过程平稳、大分子的分子量在适宜的范围内。当反应温度过低时,体系中的初级自由基浓度过低,聚合反应速率很慢,甚至难以发生;当反应温度过高时,体系中的初级自由基浓度过高,聚合过程剧烈,产物分子量不高[16]。由图4可知,反应温度为75 ℃时,产物的降黏率和反应产率较好。
2.5 反应时间优选
n(VP)=0.25 mol,n(ST)=0.3 mol,n(MA)=0.1 mol,n(SA)=1 mol,m(单体)∶m(甲苯)=3∶7,m(BPO)∶m(单体)=0.3%,反应温度为75 ℃,考察反应时间对产物降黏性能和反应产率的影响,见图5。

t/h图5 反应时间对产物降黏率和反应产率的影响
由图5可知,当反应时间从4 h延长到6 h,产物降黏率和反应产率出现明显提升,进一步增加反应时间,2个参数基本不变。因此,最优反应时间为6 h。将最优时间下制备的样品命名为PVS,待下一步分析和性能评价。
3 降黏剂表征及性能评价
3.1 降黏剂表征
通过红外光谱表征了PVS的分子结构,结果见图6。

σ/cm-1图6 PVS的红外分析结果
由图6可知,2 918、2 845 cm-1为烷基上C—H的吸收峰,来自分子主链和SA单元侧基上甲基、亚甲基和次甲基;1 846、1 783 cm-1为环状酸酐上羰基的吸收峰,来自MA单元;1 732 cm-1为酯类羰基的吸收峰,来自SA单元;1 604、1 465 cm-1为苯环骨架振动吸收峰,来自ST单元;1 645、1 415 cm-1为吡啶环骨架振动吸收峰,来自VP单元。由红外光谱可以看出,PVS为4种单体的共聚物,达到了预期的分子结构。
3.2 耐温性能评价
稠油降黏剂时常和热力采油一起使用,其耐温性能对于矿藏应用十分重要。将自制PVS和HYR分别放置在高温高压反应釜中,在不同温度下老化6 h,测定老化后2种降黏剂的降黏性能,对比分析二者的耐温性能,见图7。
由图7可知,同等条件下,PVS的降黏性能优于HYR。PVS在低于300 ℃老化,降黏性能比较稳定,降黏率超过80%,超过300 ℃后降黏性能明显下降。HYR的耐温性能不如PVS,当老化温度超过250 ℃时,降黏率显著下降。
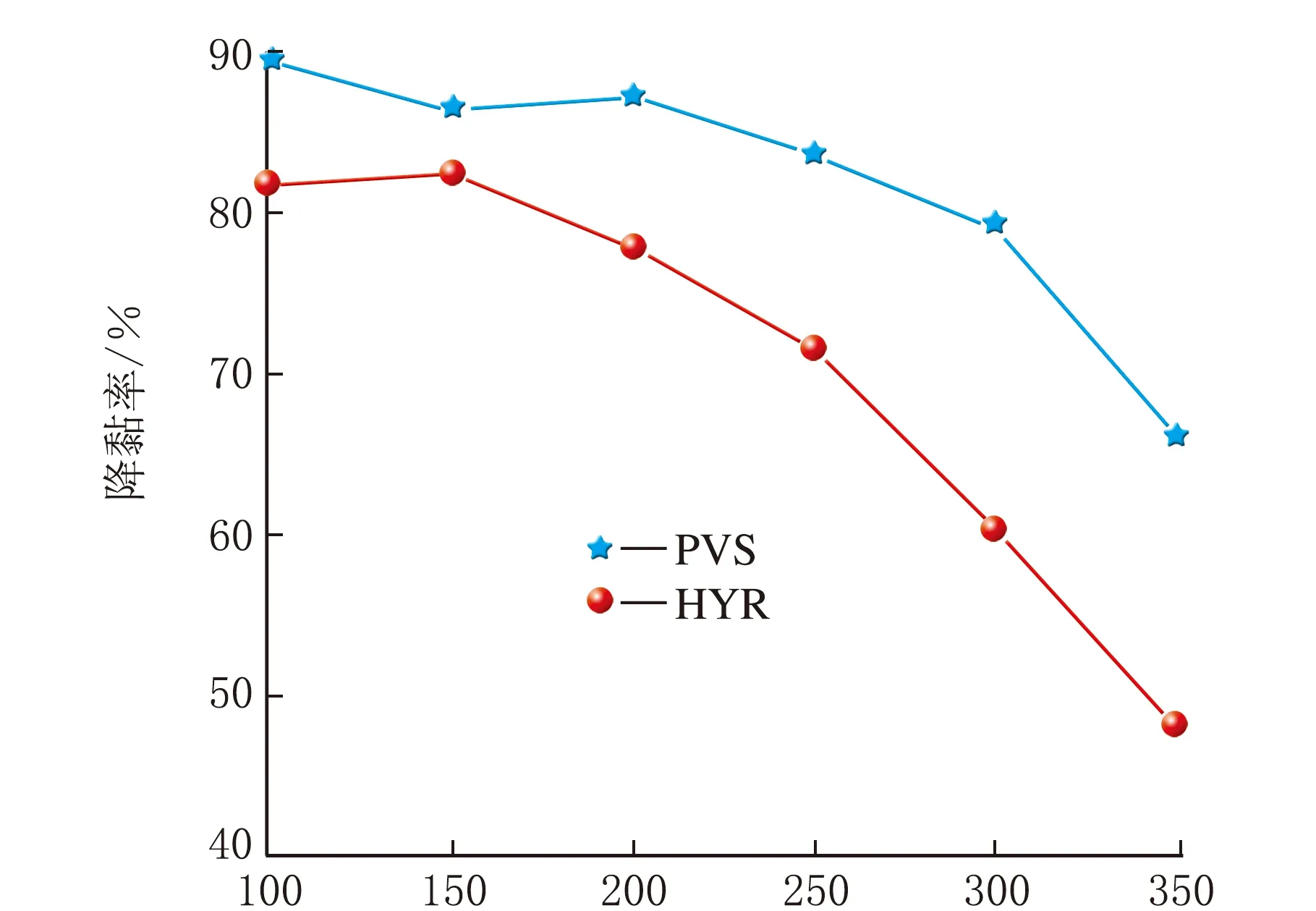
t/℃图7 老化温度对降黏率的影响
评价了PVS和HYR在300 ℃老化不同时间后的降黏性能,见图8。

t/h图8 老化时间对降黏率的影响
由图8可知,对于PVS而言,当老化时间为3~12 h,降黏性能变化不大;老化12 h,降黏率为76.9%;当老化时间超过12 h后,降黏性能显著下降;老化18 h后,降黏率为68.3%。对于HYR而言,当老化时间为3~12 h,降黏率下降明显;进一步延长老化时间,降黏率趋向于稳定;老化18 h后,降黏率为49.2%。由此可见,PVS具有更好的抗高温老化能力。
3.3 降黏效果稳定性测试
在油溶性降黏剂种类筛选和性能评测过程中,发现对于某些工业降黏剂产品,将其加入到原油中降低原油黏度后,原油黏度随着放置时间的延长而持续上升,这可能是原油中被降黏剂分子拆散的胶质、沥青质具有恢复层状重叠堆积结构的趋势,与之作用的降黏剂分子逐渐脱离,原油逐渐恢复原有的空间结构。测试了50 ℃下,加入PVS或HYR的原油在放置不同时间后的黏度,计算降黏率,见图9。

t/h图9 测试时间对降黏率的影响
由图9可知,随着时间的增加,2个原油体系的黏度均呈现上升的趋势。在放置的初期,黏度增加的速率较大,随后增加速度放缓,黏度逐渐接近一个平衡值。HYR体系的黏度增加值显著高于PVS体系,放置120 h后,PVS体系的降黏率是82.9%,高于HYR体系的69.8%。由此可见,作者研发的降黏剂具有优良的长期降黏性能。
4 结 论
(1)以VP、ST、MA、SA为单体,过氧化苯甲酰为引发剂,甲苯为溶剂,通过自由基聚合,合成了一种含有吡啶基的油溶性聚合物,该聚合物具有降低稠油黏度的作用;
(2)通过正交实验和单因素实验,确定了降黏剂最佳的合成条件为n(VP)=0.25 mol,n(ST)=0.3 mol,n(MA)=0.1 mol,n(SA)=1 mol,m(单体)∶m(甲苯)=3∶7,m(BPO)∶m(单体)=0.3%,反应温度为75 ℃,反应时间为6 h;
(3)性能评价实验表明,在w(降黏剂)=0.1%、测试温度为50 ℃,合成降黏剂的降黏率为82.9%,降黏性能优于现场用产品,此外,合成降黏剂具有良好的耐温性能,稳定的降黏性能。
