甲基丙烯酸甲酯-丙烯酸甲酯共聚物的二维核磁共振光谱分析
2020-09-10李秀萍于海波
王 博,王 旭,于 雪,李秀萍,于海波
(中国石油吉林石化公司 研究院,吉林 吉林 132021)
聚甲基丙烯酸甲酯(PMMA)的工业生产技术日趋成熟,市场前景广阔[1-4],而核磁共振光谱技术作为聚合物微观结构分析的一种重要手段,在PMMA的立构组成、序列分布、聚合机理、链增长方式等分析领域取得了较大进展[5-6],对于PMMA的衍生产品[7-8],如本研究中的甲基丙烯酸甲酯(MMA)-丙烯酸甲酯(MA)共聚物(co-MMA-MA),其单体分布及其含量直接影响着材料的规整度和加工性能。Yongsin Kim等[9]通过一维核磁共振(1D-NMR)技术中的DEPT方法对co-MMA-MA的序列分布、原料转化率和反应活性等进行了研究,但对于反映MA单体的关键谱峰归属和共聚物结合单体含量计算方法并没有给出具体说明,目前国内对co-MMA-MA的微观结构研究尚处于空白。本文综合分析了co-MMA-MA的一维核磁谱图(13C-NMR谱、DEPT谱和1H-NMR谱)和二维相关谱图(13C-1H HSQC谱和13C-1H HMBC谱),对共聚物的谱峰归属进行了详细探讨,提出了一种计算聚合物链段上MMA和MA单体含量的简便方法。
1 实验部分
1.1 原料
氘代氯仿(CDCl3)溶剂:同位素质量分数为99.8%,内标物四甲基硅烷(TMS)体积分数为0.03%,美国CIL公司;co-MMA-MA样品: MMA和MA投料质量比为9∶1,中国石油吉林石化公司研究院。
1.2 仪器及设备
AVANCE Ⅲ 400M型核磁共振波谱仪:德国布鲁克公司;核磁专用样品管:直径为5 mm,美国Norell公司;电吹风:输入功率为1 600 W,飞利浦公司。
1.3 实验条件
工作频率为400.13 MHz(1H)、100.62 MHz(13C);脉冲序列为zg(1H-NMR)、zgig(13C-NMR)、deptsp135(DEPT135)、hsqcedetgp(HSQC)、hmbcetgpl3nd(HMBC);探头为5 mm PABBO BB探头;实验温度为25 ℃。
1.4 实验步骤
(1)样品前处理:取约80 mg共聚物样品,放入核磁专用样品管中,加入0.6 mL CDCl3溶剂,用电吹风略微加热、摇动,样品即充分溶解。
(2)进样调谐:将待测样品放入磁体内,atma调谐、topshim匀场,数据点64 K。
(3)1H-NMR测试:采用脉冲序列zg,弛豫时间(D1)为4 s,累加测量16次,得到谱图后开展分析。
(4)13C-NMR测试:采用脉冲序列zgig,弛豫时间为2 s,累加测量8 000次,得到谱图后开展分析。
(5)DEPT135测试:采用脉冲序列deptsp135,弛豫时间为2 s,累加测量1 024次,得到谱图后开展分析。
(6)13C-1H HSQC测试:采用脉冲序列hsqcedetgp,弛豫时间为2 s,累加测量2次,得到谱图后开展分析。
(7)13C-1H HMBC测试:采用脉冲序列hmbcetgpl3nd,弛豫时间为2 s,累加测量8次,得到谱图后开展分析。
2 结果与讨论
2.1 谱图分析
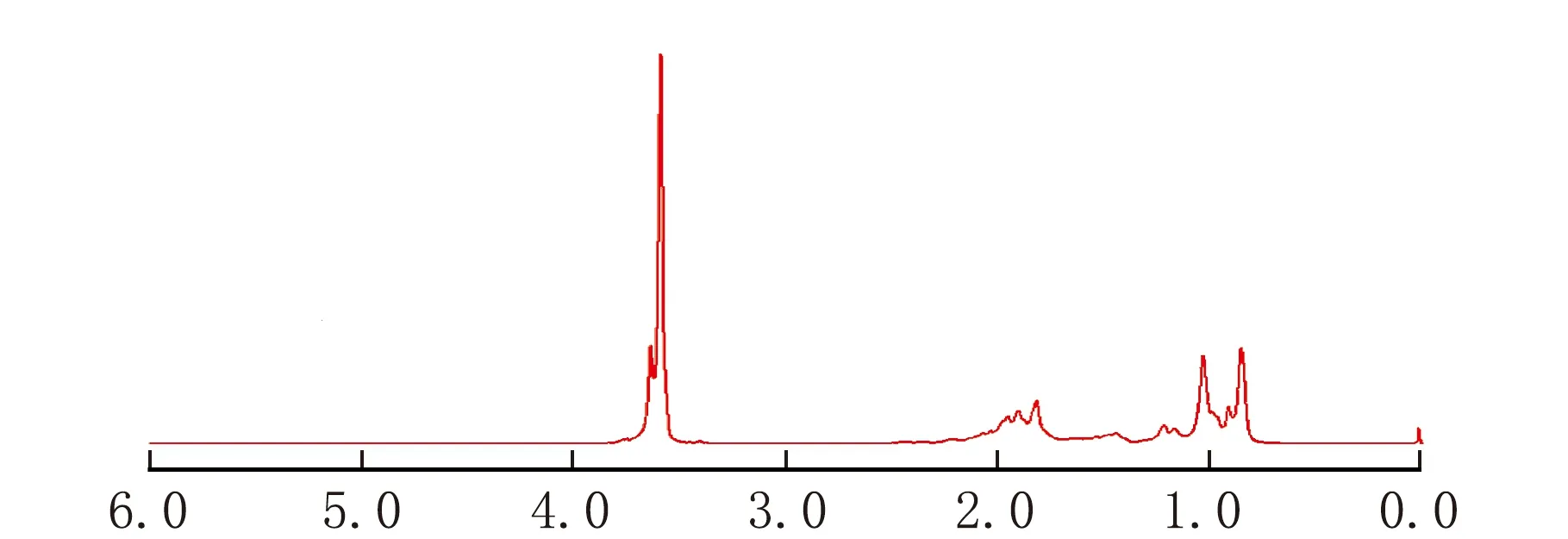
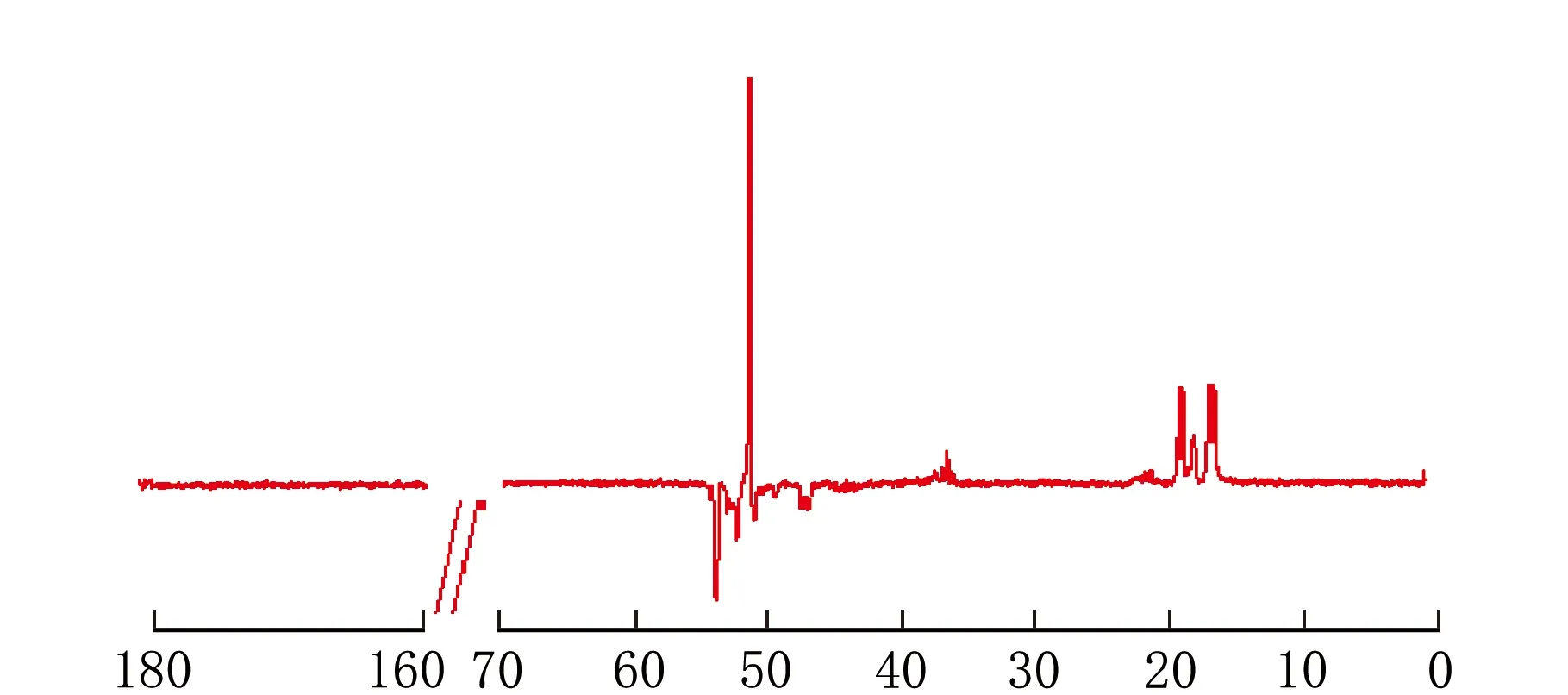
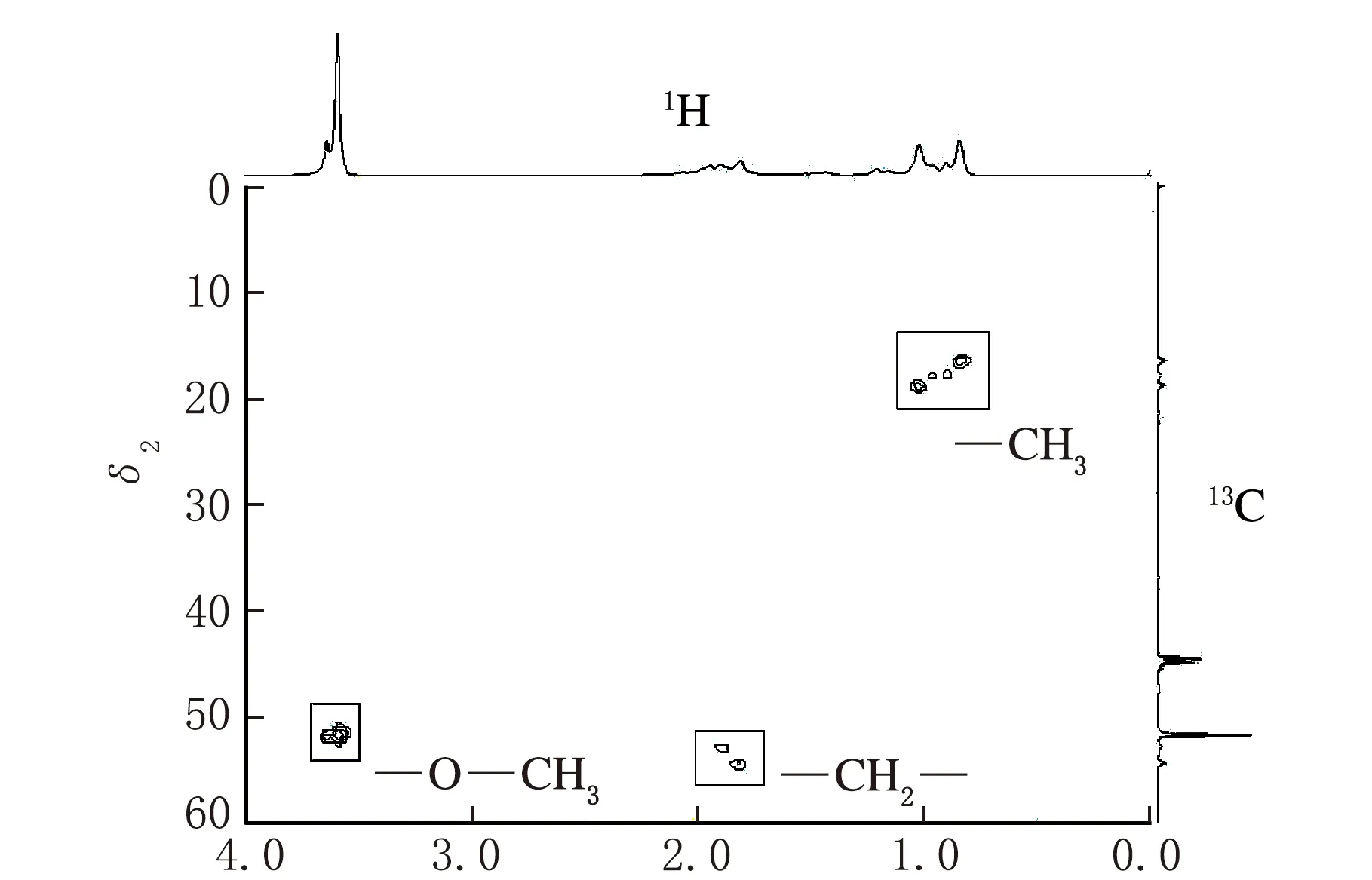
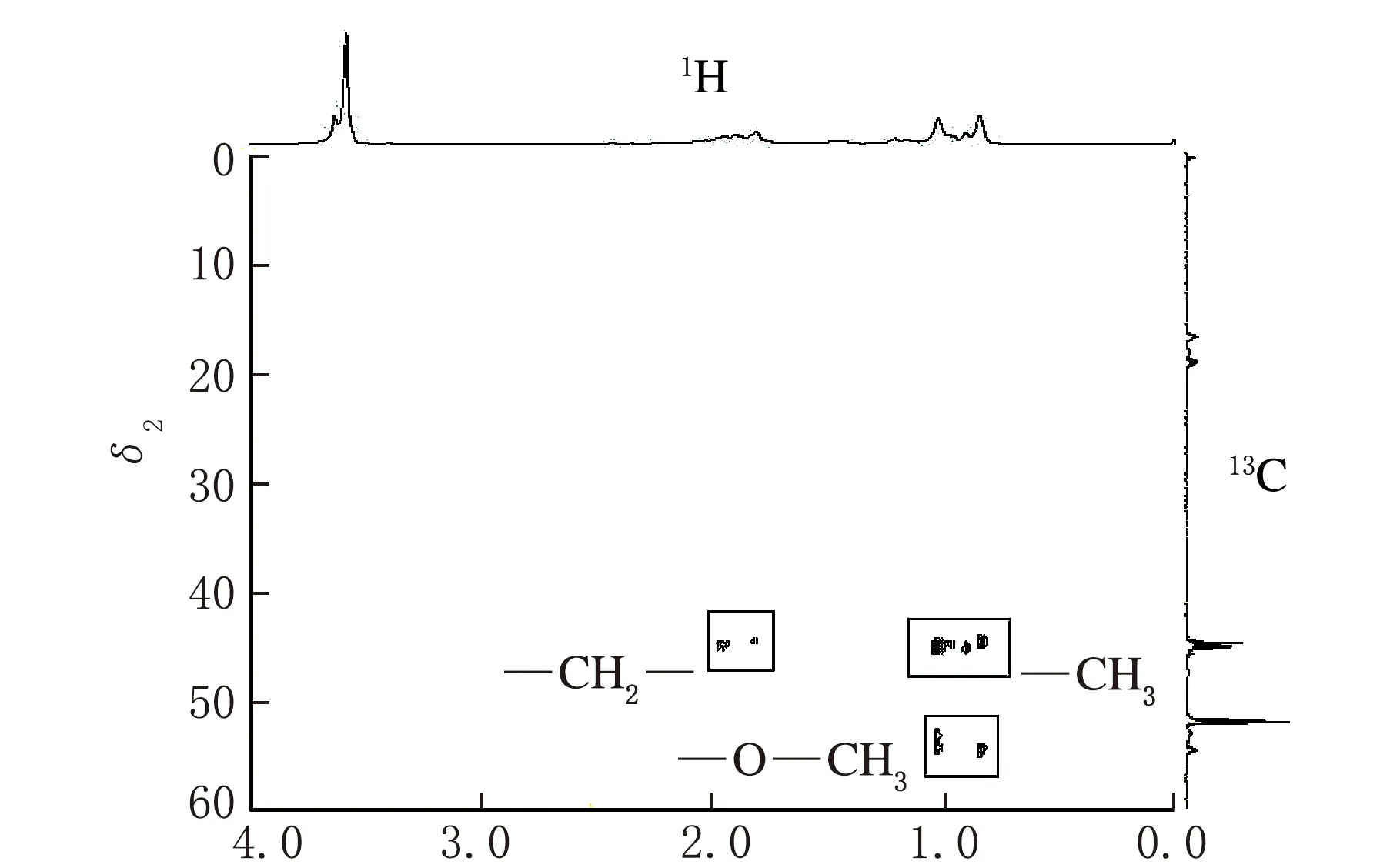
依据上述条件测定co-MMA-MA样品,得到图1所示的1H-NMR谱图、图2所示的13C-NMR谱图、图3所示的DEPT135谱图、图4所示的HSQC二维相关谱图和图5所示的HMBC二维相关谱图。其中,一维谱图的横坐标表示1H核或13C核的化学位移值,以δ表示。二维谱图上方横轴呈现1H-NMR谱图,右侧纵轴呈现13C-NMR谱图,下方横坐标表示1H核的化学位移值,以δ1表示,左侧纵坐标表示13C核的化学位移值,以δ2表示。
由图1可以看出,区域A(δ在0.6~1.4的区间)、区域B(δ在1.4~2.5的区间)和区域C(δ在3.3~3.9的区间)的积分面积比约为3∶2∶3,出峰位置及峰强度与文献中的PMMA出峰情况类似[10],可以推测该聚合物的主体为类PMMA结构。由于O原子的电负性强,影响临近的甲基质子峰向低场方向移动,因此A、B和C的积分面积大体上对应于链段中的甲氧基(—O—CH3)、亚甲基(—CH2—)和甲基(—CH3)上的质子数量。
δ
由图2和图3可以看出,仲碳原子对应于图2中δ在52~54和47~48区间的三组峰,季碳原子为δ在44~46区间的三组裂分峰,而δ在36~38区间的多重峰则包含了不同立构下的叔碳或伯碳原子峰。
δ
结合HSQC二维相关谱图(见图4),可以确定甲氧基上的伯碳原子(对应于图2中δ=51.74)和甲基上的伯碳原子(由δ在15~20区间三组峰可知)的存在。由图4(b)HSQC局部放大图和图5可知,碳谱上δ在36~38区间的多重峰仅为不同立构下的叔碳原子峰,这也反映了MA单体在链段上呈随机分布状态。

δ
δ1(a) 整体
δ1
如果忽略少量的MA对MMA聚合链段立构的影响,则可以认为伯碳原子区反映了链段整体的立构差异,也就是说,考虑到羰基氧原子电负性对临近碳核的影响,推测δ在21.05~21.50处的伯碳反映了链段的间同立构(rr)情况,δ在17.74~19.01处的伯碳反映了链段的无规立构(mr)情况,而δ在16.34处的伯碳则反映了链段的全同立构(mm)情况,如图6所示。
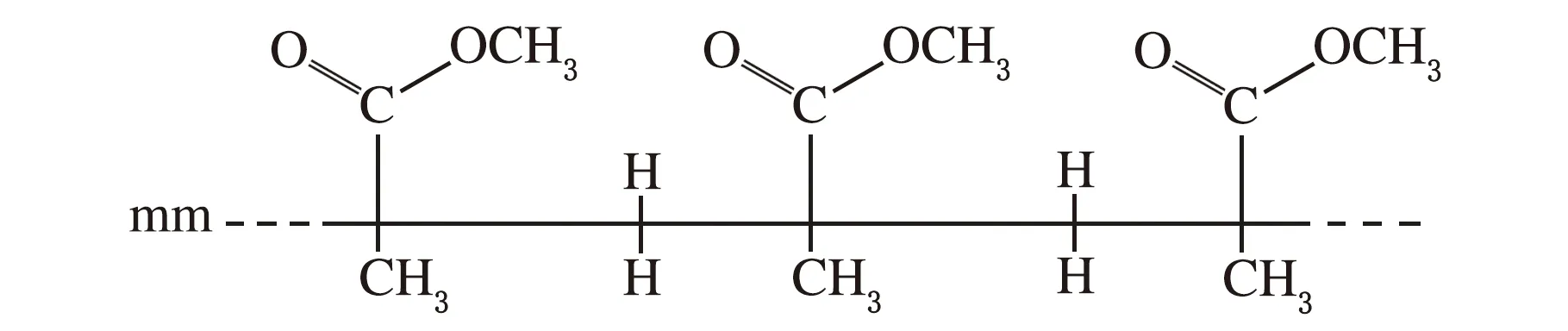
(a) 全同立构
根据以上讨论,可对co-MMA-MA氢谱和碳谱的主要谱峰进行归属,列于表1中。

表1 co-MMA-MA的谱峰归属
2.2 单体含量计算
2.2.1 碳谱法
前面提到,碳谱上δ在36~38的多重峰为叔碳原子峰,而叔碳原子只可能存在于链段上的MA单体中,因此该多重峰的面积值(设为x)正比于MA的单体含量。此外,由于δ在15~20的三组峰为伯碳原子峰,而伯碳原子只可能存在于MMA单体中,因此伯碳区的面积值(设为y)正比于MMA的单体含量。MA单体摩尔分数n1的计算如式(1)所示,MMA单体摩尔分数(m1)的计算如式(2)所示。
(1)
(2)
2.2.2 氢谱法
由图4可知,碳谱上δ为36~38区间的叔碳峰对应于氢谱上δ为2.18~2.30的叔氢峰,因此氢谱上叔氢峰的面积值(设为x′)正比于MA的单体含量,氢谱上δ为0.6~1.4区间的伯氢峰的面积值(设为y′)正比于MMA的单体含量的3倍。MA单体摩尔分数(n2)的计算如式(3)所示,MMA单体摩尔分数(m2)的计算如式(4)所示。
(3)
(4)
2.2.3 精密度结果
用1H-NMR对同一样品测定6次,结果保留1位小数,精密度结果见表2。相对标准偏差结果表明该方法测定结果的精密度符合要求。
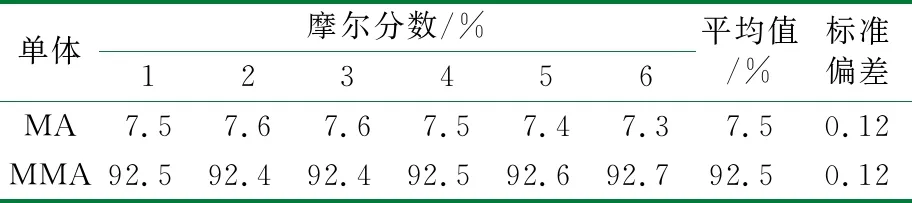
表2 1H-NMR测定的精密度
用13C-NMR对同一样品测定6次,结果保留1位小数,精密度结果见表3。相对标准偏差结果表明该方法测定结果的精密度符合要求。
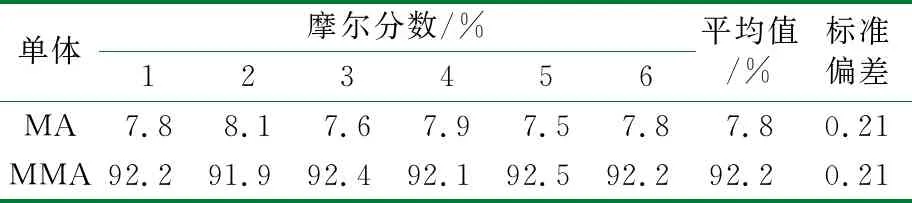
表3 13C-NMR测定的精密度
2.2.4 两种方法计算结果的差异
将利用1H-NMR和13C-NMR测得的单体含量数据(即多次测定摩尔分数平均值)进行比较,结果列于表4中。

表4 1H-NMR和13C-NMR测定结果平均值比较
由表4可知,两种方法得到的结果基本相等,符合理论预期。由于氢谱相对于碳谱具有简便易行、信噪比高、省时高效(氢谱只需要扫描几分钟,碳谱往往需要扫描2 h以上)等优点,因此在日常检测中只需要开展氢谱实验就可以得到较为满意的结果。
3 结 论
(1)在一维核磁共振波谱的基础上,利用二维核磁共振波谱法,任何共聚比的co-MMA-MA的谱峰归属都可得到详细说明。
(2)利用核磁共振氢谱技术,大批量co-MMA-MA样品的结合单体含量可以简便、快捷、准确地计算出来,这对于产品研发阶段工艺技术的持续改进和工厂生产阶段的质量控制有极强的应用价值。
