过渡金属催化的不对称芳基-芳基偶联反应:轴手性联芳基化合物的合成研究进展
2020-09-09刘慧慧杜思思梁娱婧张安安
刘慧慧,杜思思,梁娱婧,周 倩,王 露,吴 韦,张安安*
(1.东北石油大学 研究生院,黑龙江 大庆 163000;2.商丘师范学院 化学化工学院,河南 商丘 476000)
轴手性联芳类结构是天然产物中常见的结构单元,这些天然产物及其异构体大多具有良好的生理活性(A,Chart 1)[1-3]。在不对称合成领域,轴手性的联芳类化合物是一类重要的手性配体或催化剂,具有优秀的活性和选择性,其催化的不对称反应已经应用于药物的工业生产,成为不对称合成中的明星分子(B,C,Chart 1)[4-6]。
轴手性联芳基化合物重要的应用引起了化学工作者对其不对称合成方法研究的极大兴趣。Hayashi小组[7]报道的镍催化不对称 Kumada-Tamao-Corriu偶联反应拉开了过渡金属催化芳基-芳基不对称偶联合成轴手性联芳类化合物的帷幕。30多年来,合成化学家们在反应体系的开发、催化剂的设计方面进行持续地研究,取得了重要进展,发展诸如过渡金属催化的芳基-芳基的不对称偶联、芳香环的构建、中心手性向轴手性的传递、联芳基化合物的去对称化、不对称碳氢活化、环状化合物的开环-偶联等多种合成方法。作为较早发展的策略,过渡金属催化的芳基-芳基的不对称偶联具有通用性好、原子经济性高、官能团兼容性强等优点。本文按过渡金属催化剂分类介绍芳基金属和芳基亲电试剂的交叉偶联反应(包括Negishi偶联、Suzuki偶联)、富电子芳基化合物的氧化偶联反应在轴手性联芳类化合物合成中的研究进展(Chart 2)。
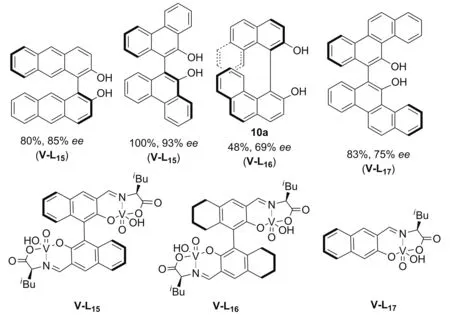
Chart 3

Chart 1
Chart 2
1 镍催化的不对称芳基-芳基偶联反应
1988年,Hayashi与Ito小组[7]采用具有面手性和中心手性的二茂铁单膦配体L1、L2率先实现镍催化2-烷基-1-萘溴化镁与1-溴萘衍生物的不对称交叉偶联反应,产物2,2′-二甲基-1,1′-联萘的ee值可达95%。而不带中心手性的配体L3只能给出相应的外消旋产物(Scheme 1),说明中心手性和面手性的协同效应及烷氧基的辅助配位作用对反应的选择性起至关重要的作用。

Scheme 1
Scheme 2
Scheme 3

Scheme 4

Scheme 5
Scheme 6
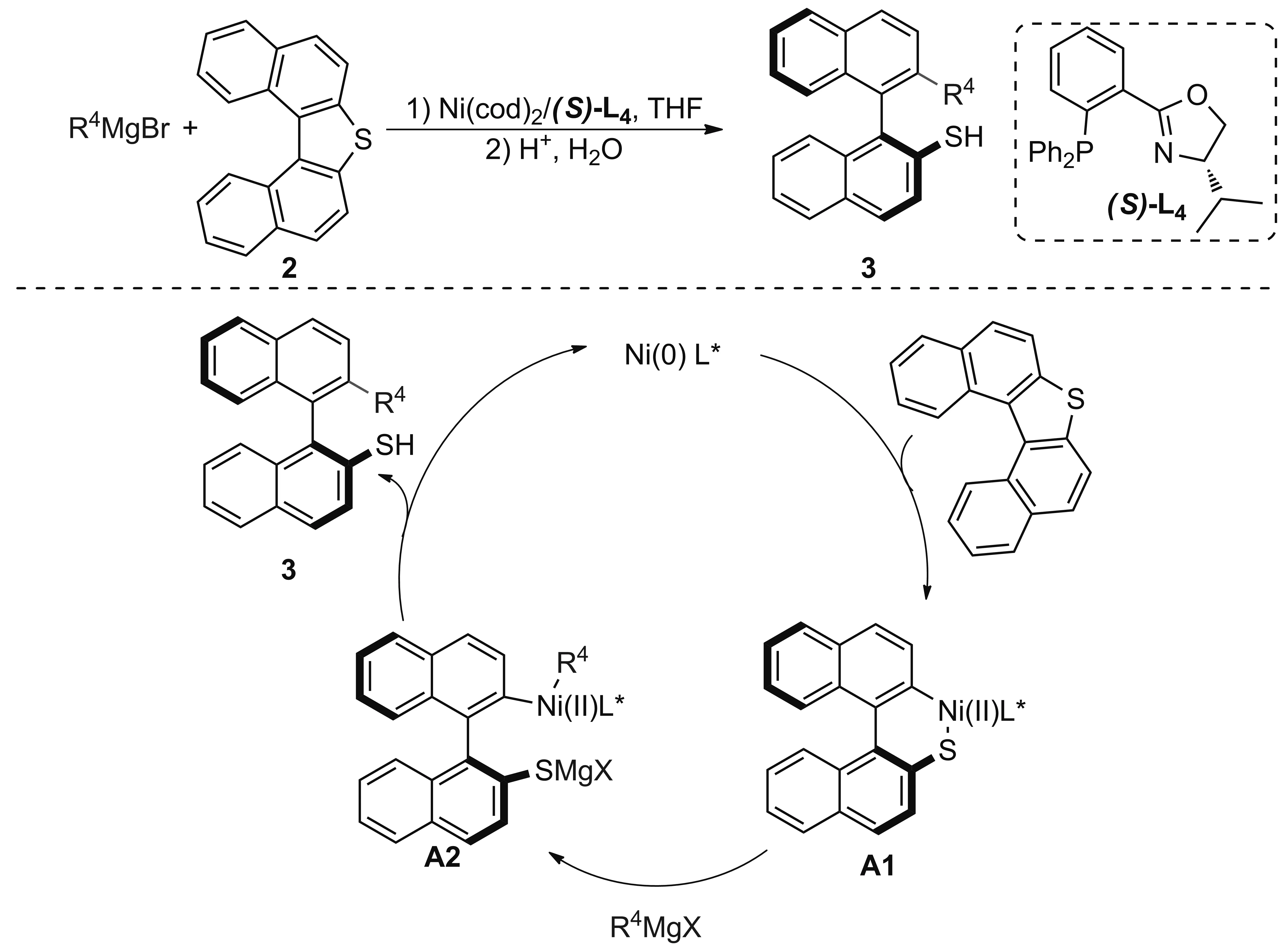
2002年,Hayashi小组[8]又利用Ni(cod)2/(S)-L4催化体系实现了芳基格氏试剂与二萘并噻吩的开环交叉偶联反应,2′-芳基-2-硫醇联萘产物ee值高达95%。反应的机理如下:首先,二萘并噻吩底物与零价镍的络合物发生氧化加成生成六元的二价镍杂环A1,A1与格氏试剂的转金属化反应生成中间体A2,后者发生还原消除生成最终产物3,同时完成催化剂的再生(Scheme 2)。
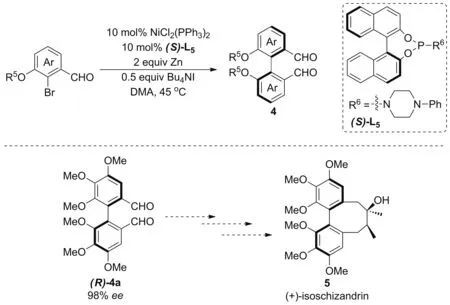
2010年,林国强与徐明华课题组[9]实现2-溴-3-烷氧基芳醛衍生物的不对称Ullmann偶联。反应以联萘骨架的手性亚膦酰胺(S)-L5为配体、NiCl2(PPh3)2为催化剂,收获了中等收率与ee值。偶联产物(R)-4a经五步反应即可得到天然产物(+)-异五味子素(5,Scheme 3)。
2 铜催化的不对称芳基-芳基氧化偶联反应
Wynberg[10],Brussee[11-12],Yamamoto[13-14]和Kocovsk等[15-16]开创了苯酚、萘酚的不对称氧化偶联,发现化学计量的手性胺/铜(Ⅱ)配合物能催化官能团化手性BINOLs的合成。Nakajima将氯化亚铜与L-脯氨酸衍生的二胺L6用于2-羟基-4-甲酸苄酯萘的催化不对称氧化偶联[17](a,Scheme 4),Kozlowski发展的顺式-1,5-二氮十氢化萘配体(S,S)-L7具有更高的对映选择性,ee值高达94%(b,Scheme 4)[18-19]。该催化体系实现了(+)-Phleichrome、(+)-Calphostin D[20]、Cercosporin[21]、Hypocrellin A[22]、(S)-Bisoranjidiol[23]等天然产物的合成。Gao课题组[24]尝试大环双铜(II)配合物Cu-L8催化2-萘酚的不对称氧化偶联,反应条件温和、产率和对映选择性好(c,Scheme 4)。
3 钒催化的不对称芳基-芳基氧化偶联反应
2001年,Chen小组和Uang小组[25-26]分别由手性氨基酸与水杨醛衍生物合成了席夫碱三齿配体,与VOSO4反应可以高产率的生成手性单核氧钒(IV)配合物V-L9、V-L10,它们在反应中能给出中等的对映选择性(Scheme 5)。Gong课题组[27]发现具有轴手性和中心手性的联萘酚骨架的双核氧钒配合物V-L11能够给出高达98%的ee值。Sasai小组[28]将V-L11中的两个金属中心之间的氧桥(V—O—V键)打开,得到具有两个相同金属催化中心的双核氧钒(IV)配合物V-L12。相较于单核氧钒配合物,双核氧钒配合物V-L12能使2-萘酚衍生物7的不对称偶联速度提高30倍、条件更温和,底物范围更广;值得一提的是,空气可以代替氧气作为反应的氧化剂(Scheme 5)。
Scheme 7
Gong和Luo[29]进一步发展了联芳类双核氧钒(V)配合物(V-L13,V-L14,Scheme 5)。在空气氛围下,5 mol%的V-L13能顺利催化2-萘酚的不对称氧化偶联,ee值高达97%。有意思的是,在非手性联苯酚和手性氨基酸组成的催化剂V-L14中,氨基酸的中心手性能够有效的传递到联苯酚的轴手性,催化2-萘酚衍生物8的不对称氧化偶联反应,以定量的产率和高达98%的ee值生成一系列手性联萘酚衍生物。
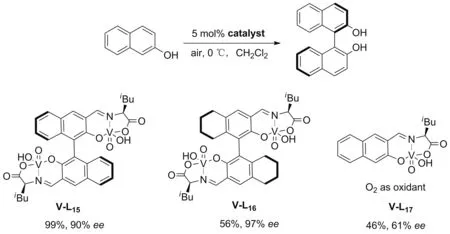
偶然的发现使Sasai小组[30]以VOCl3为原料高效地合成了五价钒催化剂V-L15、V-L16,V-L17。双核催化剂V-L15与V-L16在萘酚的氧化偶联中能给出优秀的对映选择性;而单核催化剂V-L17的催化活性和选择性都不理想。以上结果再次证实了双核催化剂的协同作用(Scheme 6)。
Scheme 9
Scheme 8
2014年,Sasai[31]将双核氧钒配合物V-L15、V-L16、单核氧钒配合物V-L17成功用于联稠环二酚的对映选择性合成,但偶联产物10a的产率明显较低(Chart 3)。通过稠环酚原料的交叉偶联实验和前期相关机理研究,作者认为菲-3-醇9a的中间体稳定性过高,是导致其转化率低的原因。
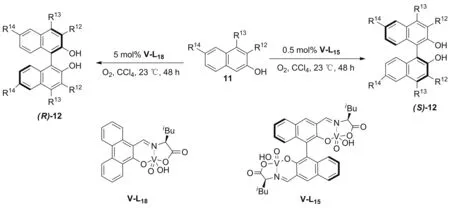
2017年,Takizawa和Oh[32]小组在2-萘酚衍生物11的不对称偶联反应中,发现单核氧钒V-L18催化生成产物(R)-12,而以双核氧钒V-L15为催化剂时,生成构型相反的产物(S)-12(Scheme 7)。另外,由于协同效应双核催化剂V-L15对映选择性和反应活性都优于单核催化剂V-L18。以4-取代-2-萘酚的氧化偶联为模型,探讨了这两种氧钒催化剂的作用机理。通过不同氧化电位的4-取代-2-萘酚的交叉偶联实验,发现双核氧钒V-L15的催化过程既不符合自由基-自由基,也不符合自由基-阴离子偶联机制,而最可能是V-L15的两个钒中心与两分子4-取代-2-萘酚配位后发生分子内偶联。
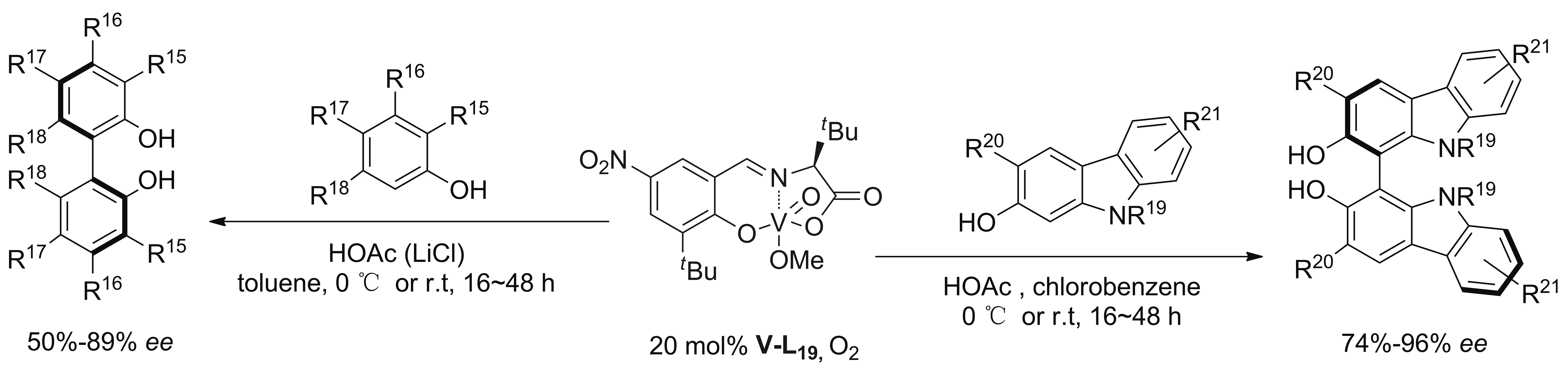
较2-萘酚类底物而言,2-苯酚类底物较难氧化,反应位点复杂,区域选择性更难控制。2017年,Kozlowski课题组[33]首次将单核氧钒配合物V-L19与路易斯酸或LiCl结合实现2-苯酚类、2-羟基-咔唑类底物的不对称氧化偶联(Scheme 8)。LiCl或路易斯酸在2-苯酚类底物的不对称氧化偶联中起关键作用,实验和DFT计算结果表明,反应中形成了氧钒2-苯酚类的二聚体,LiCl或路易斯酸的加入加速了这一过程[34]。
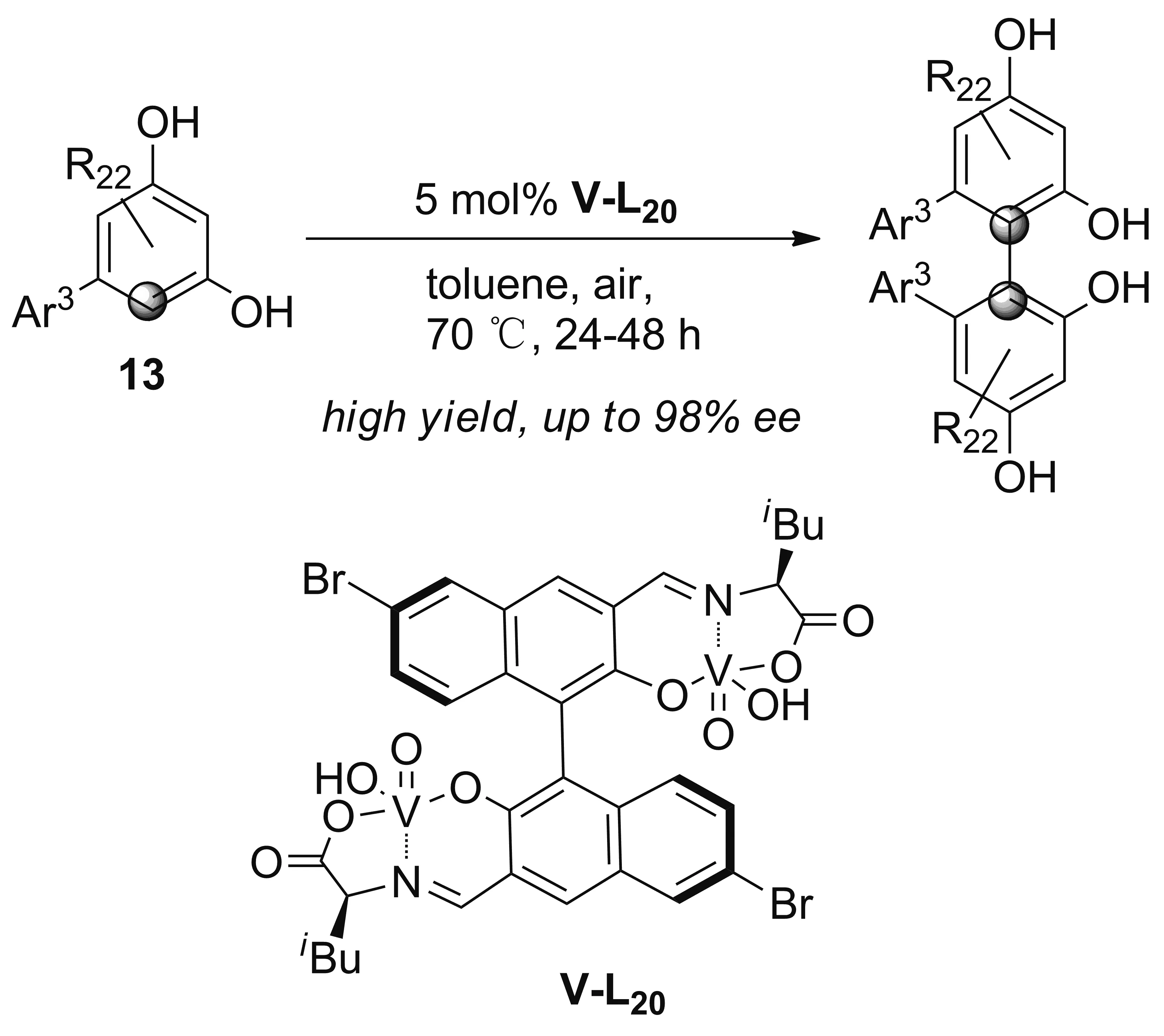
由于反应位点多,间苯二酚类化合物的区域选择性和对映选择性氧化偶联反应存在很大的挑战。2018年,Sasai和Takizawa小组[35]对其之前开发的双核氧钒V-L15中联萘骨架的6,6’-位进行溴化。新双核氧钒配合物V-L20在间二苯酚类化合物13的不对称氧化偶联中,区域选择性好、产率高、对映选择性高达98%,进一步丰富苯酚类化合物的不对称氧化偶联反应(Scheme 9)。
铁催化酚类氧化偶联反应是制备复杂双芳基酚类骨架的有效方法。2009年,Katsuki和Egami首次在不使用添加剂的情况下,实现铁配合物[Fe(salan)]2催化的2-萘酚衍生物的不对称氧化偶联反应(Scheme 10)。动力学研究显示单体[Fe(salan)]可能参与反应,并通过[Fe(Salan)](3-溴-2-萘酚)单晶结构验证。最终确定[Fe(Salan)](3-取代-2-萘酚)中间体和氧气参与了铁催化不对称氧化交叉偶联的决速步骤,符合自由基-阴离子机制[36-37]。
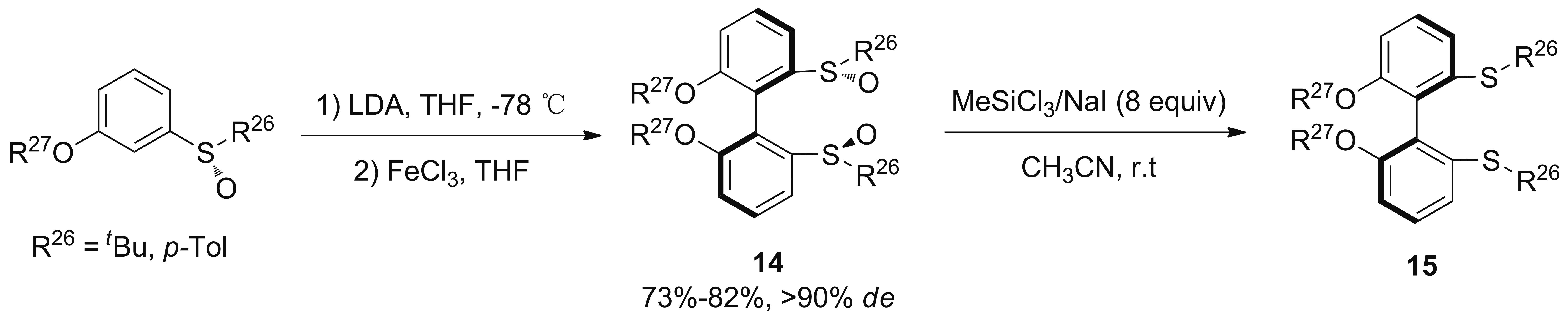
周永贵和李玉学课题组[38]以三氯化铁为氧化剂实现手性芳基亚砜的立体选择性氧化偶联,合成了轴手性双亚砜联苯配体14,de值高达90%以上(Scheme 11)。配体14在铑催化芳基硼酸与环己烯-2-酮的1,4-不对称加成中对映选择性高达99%,亦可转化为相应的硫醚配体15。
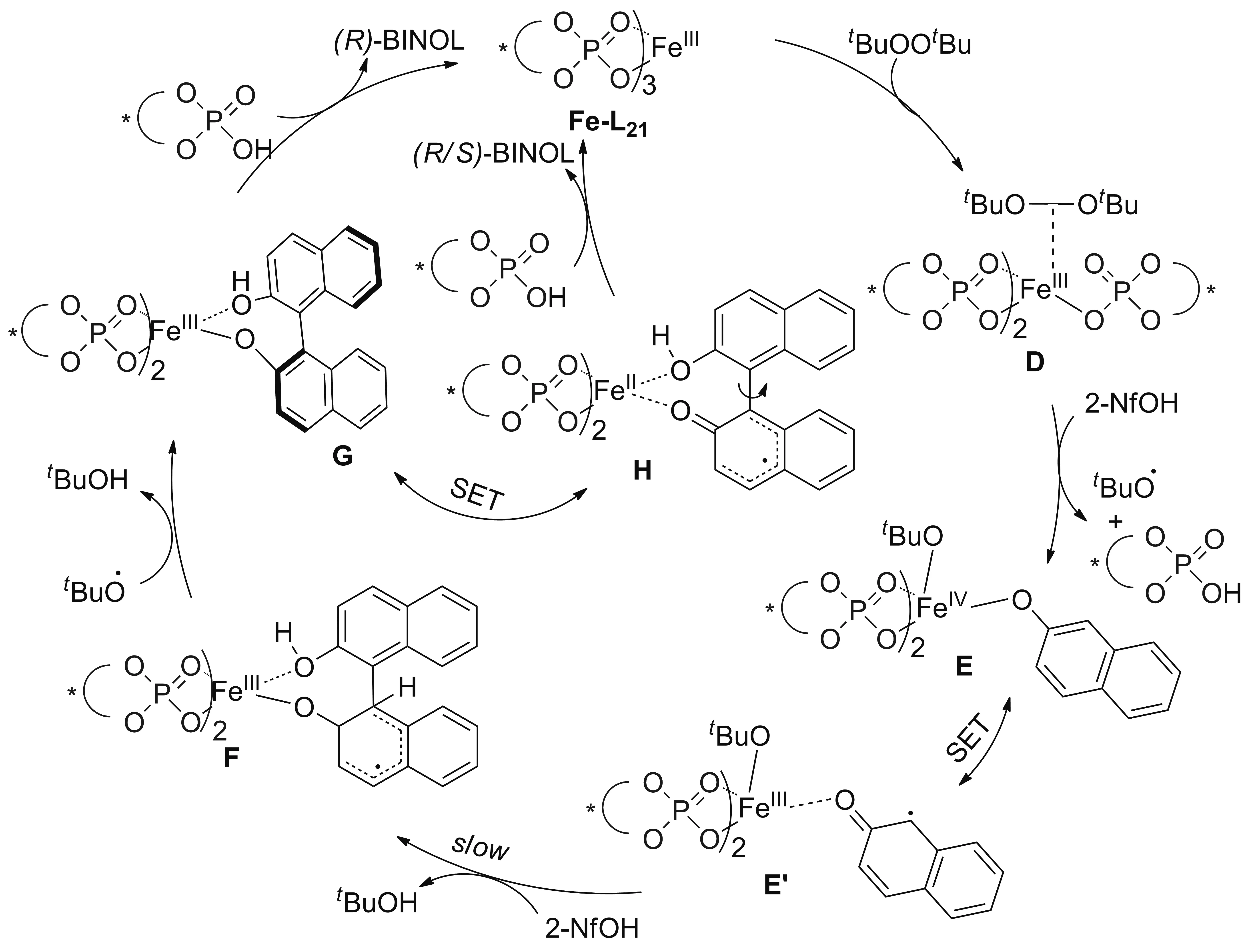
2016年,Pappo课题组[39]将手性磷酸铁配合物Fe-L21引入到2-萘酚、2-萘酚衍生物16的不对称氧化(交叉)偶联中,产率中等至良好,ee值高达92%(Scheme 12)。动力学研究符合Katski[37]提出的自由基-阴离子偶联机制,但tBuOOtBu与手性磷酸铁的配位并不慢,因此催化过程由tBuOOtBu与Fe-L21配位中间体D开始,然后过氧键裂解,2-萘酚与Fe-L21中一分子磷酸发生配位交换,生成铁(IV)中间体E,亲电的单电子转移(SET)萘氧自由基中间体E′与另一分子亲核的2-萘酚发生自由基-阴离子偶联生成G,再与手性磷酸配体交换释放轴手性联萘酚产物,而G和H之间的SET过程会导致产物的光学纯度降低(Scheme 13)。

Scheme 10
Scheme 11

Scheme 12
Scheme 13

Scheme 14
2018年,Pappo小组[40]发展了三氯化铁催化3-芳基-2-萘酚与手性萘胺19的立体选择性氧化交叉偶联,得到光学纯氧化偶联产物(Sa,S)-NOBINS。手性辅助可通过钯催化的氢化反应脱去,得到光学纯的相应产物(Sa)-20。交叉偶联产物在催化体系中存在非对映异构体相互转化现象,导致光学纯度降低;而当手性轴间位为大位阻官能团时,不容易出现此类异构现象(Scheme 14)。
5 钯催化的不对称Suzuki偶联反应
5.1 钯催化的对映选择性Suzuki偶联
2000年,Buchwald课题组[41]使用单膦配体(S)-L22用于实现钯催化的不对称Suzuki偶联,ee值高达92%(a,b,Scheme 15)。DFT计算与实验研究表明,卤代烃磷酸酯或酰胺上的氧与钯配位作用有利于高对映选择性的提高[42]。之后,Qiu课题组[43]报道了高效的手性碳桥联的联芳单膦配体L23诱导的3-溴-4-取代吡啶与2-,4-取代-1-萘硼酸的不对称Suzuki偶联(c,Scheme 15),对映选择性良好、底物范围更广。
Tang课题组[44]开发出构象良好的苯并五元氧膦膦中心手性配体L24~L26,在钯催化芳基硼酸与邻甲酰代芳基溴的不对称Suzuki偶联中具有优异的反应活性,尤其是与邻甲酰苯并噁唑啉酮芳基溴偶联时,ee值高达96%(a,Scheme 16)。经证实还原消除过程中1-萘硼酸的萘环与邻甲酰苯并噁唑啉酮溴萘的甲酰苯并噁唑啉酮存在π-π次级作用。此外,配体L24也可实现邻磷酰基芳基溴与芳基硼酸的不对称Suzuki偶联,ee值大于80%(b,Scheme 16)。

Scheme 15

Scheme 16
Suginome课题组[45-46]致力于新型手性螺旋聚合物膦配体(P)-(R)-PQXphos的开发,相继实现了芳基硼酸与邻磷酰基卤化萘、邻甲酰基卤化萘的不对称Suzuki偶联,对映选择性良好(Scheme 17)。聚合物配体L27~L29的螺手性不仅受手性末端官能团和手性侧链共同影响而且与溶剂密切相关:在1,1,2-三氯乙烷中其螺手性发生翻转,偶联产物的优势构型也随之反转。
Scheme 18

Scheme 17
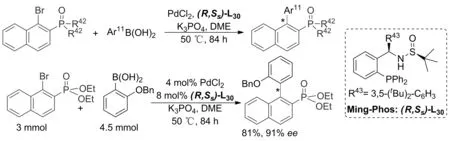
2019年,Qiu小组[47]将手性亚磺酰胺配体Ming-Phos用于钯催化邻磷酰基溴萘与芳基硼酸的不对称Suzuki偶联。利用PdCl2和(R,Ss)-L30原位形成的催化剂,成功催化22种轴手性酰基衍生物的合成,ee值高达98%。
该催化体系非常适合富电子基芳基硼酸,克级反应效果良好(Scheme 18)。
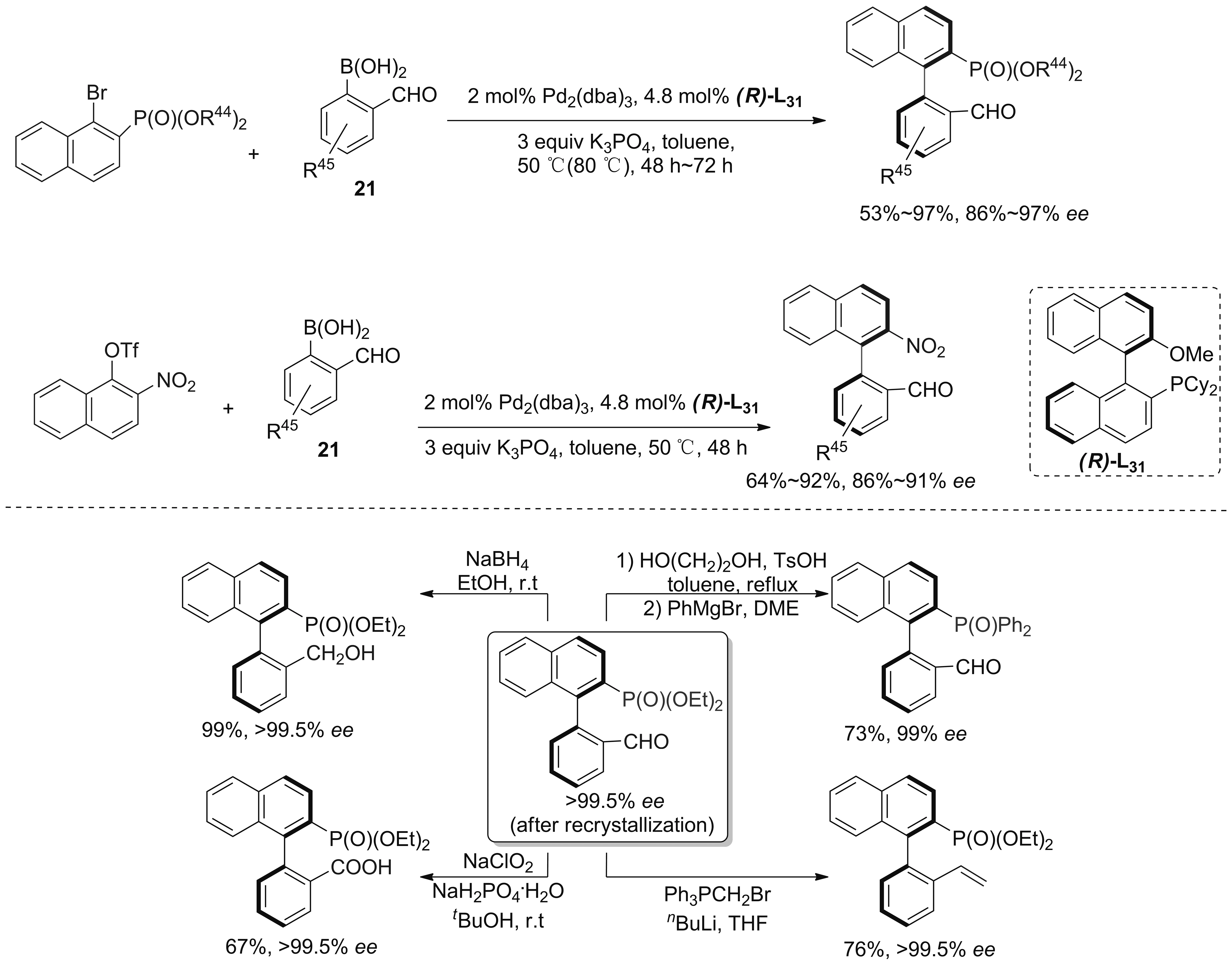
Qiu小组[48]发展了邻磷酰基溴萘、2-硝基-1-(三氟甲磺酸酯)萘与2-甲醛苯硼酸衍生物的不对称suzuki偶联,ee值为86%~97%,产物可以方便地衍生为其他轴手性化合物(Scheme 19)。
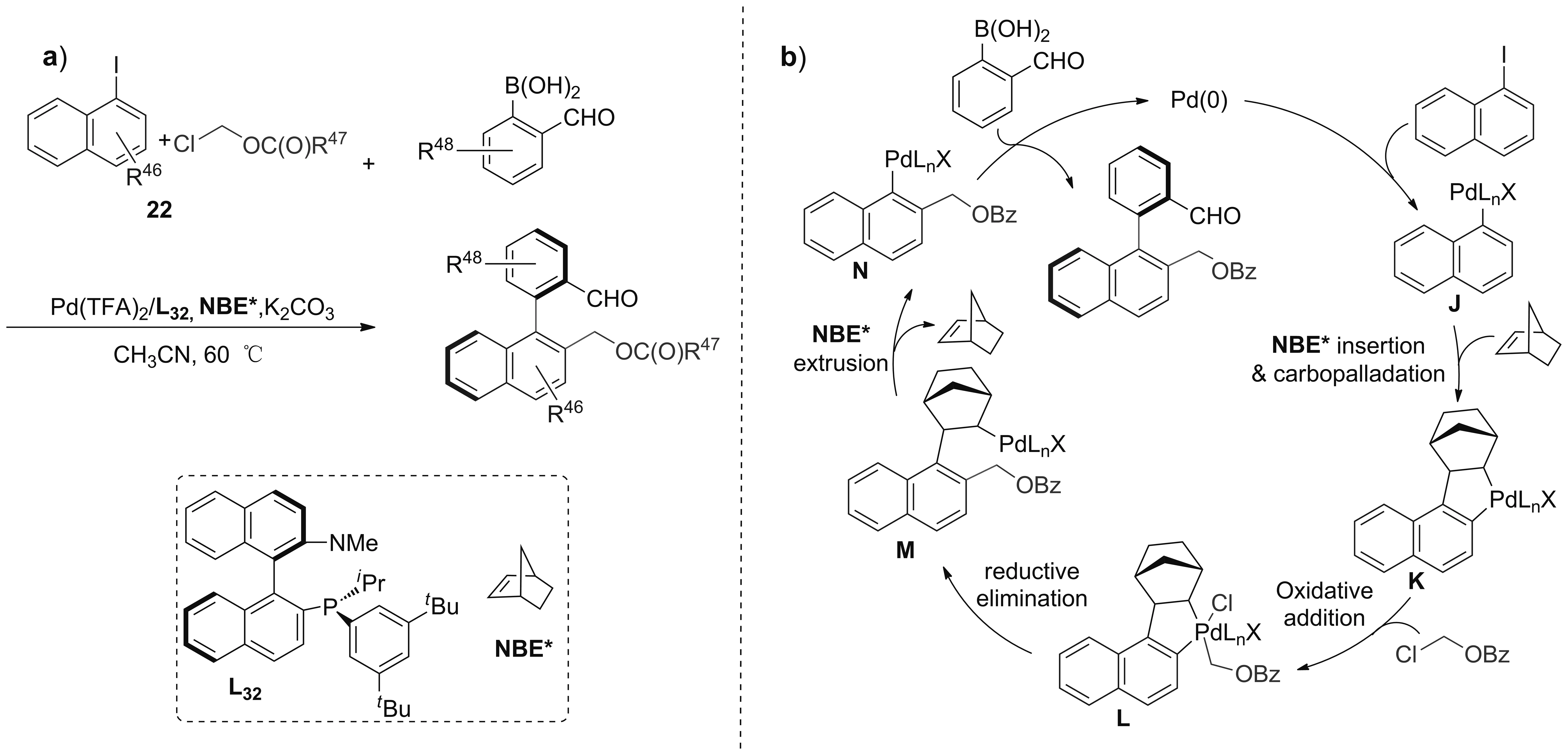
2018年,Gu课题组[49]以Pd(TFA)2/降冰片烯(NBE*)/L32/K2CO3为催化体系,完成了1-碘萘衍生物、烷(芳)甲酸-1-氯甲酯、2-甲醛苯硼酸衍生物三组分底物的不对称Suzuki偶联,对映选择性高达96%(a,Scheme 20)。作者认为反应历程可能是:Pd(0)与1-碘萘氧化加成生成萘基钯中间体J,NBE*插入C—Pd键(碳钯化)得中间体K,与亲电试剂二次氧化加成得中间体L,还原消除得C—Pd中间体M,NBE*离去生成2-取代-1-萘基钯N,最后与2-甲醛苯硼酸发生经典的不对称Suzuki偶联(b,Scheme 20)。
Tang小组尝试邻氧代芳基溴与芳基硼酸的不对称Suzuki偶联。R49为大极性的BOP基团时,配体L24′/L33/L34表现出优异的反应活性和对映选择性(a,Scheme 21),经证实还原消除过程中高度极化的OBOP基团与芳基硼酸的芳基部分存在极性-π作用。配体L34对邻磷酸二乙酯溴萘与2-甲醛苯硼酸偶联也有95%的产率和96%的ee值,L33还成功地用于轴手性天然产物Michellamine B、Korupensamines A、Korupensamines B的合成[50]。
此后,Fandrick课题组将膦中心手性配体(S)-BI-DIME用于HIV整合酶抑制剂O的高效合成[51](b,Scheme 21)。随后,2-三氟甲磺酸酯(OTf)溴萘与芳基硼酸的不对称Suzuki偶联也被开发出来(c,Scheme 21),菲、并四苯等稠环硼酸参与偶联时产率和对映选择性高达90%[52]。
Scheme 23

Scheme 22

Scheme 21
Scheme 19
Scheme 20
联芳基化合物的2-,2′-位为H、CH3、Et、OCH3、OEt、OiPr、OBn、CHO、Ph等简单基团时,由于较低的旋转位阻其轴手性控制充满挑战,Zhang[53]和Reisman[54]等综述了双腙钯、双烯钯、NHC卡宾钯等各种钯配合物以及手性膦配体参与的不对称Suzuki偶联与Negishi偶联来构建这类轴手性双芳基化合物。二茂铁单膦配体PPFA[55]、双腙钯Pd-L36[56]及两亲树脂接枝的手性单膦配体PS-PEG-L37[57]虽取得一些成效,但开发出一条对映选择性好且底物普适性好的策略仍然是困难的(Chart 4)。
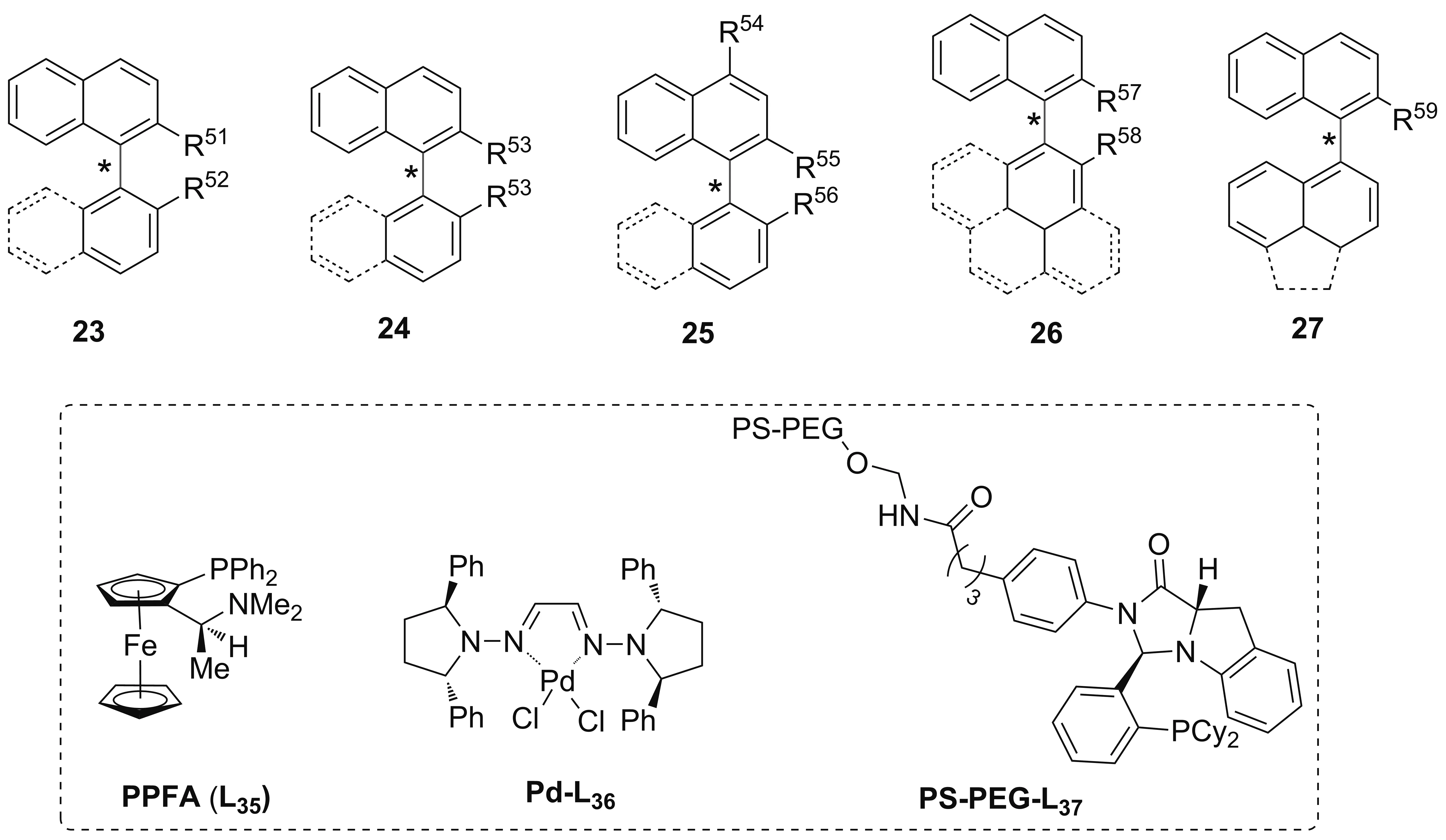
Chart 4
2018年,Senanayake和Kozlowski小组[58]尝试不对称Suzuki偶联与Negishi交叉偶联策略构建轴手性C2-对称的2,2′-二取代-1,1′-联萘化合物,取得了中等到良好的ee值(Scheme 22)。经DFT分析,Negishi交叉偶联的还原消除步骤决定对映选择性;而Suzuki偶联的选择性受氧化加成、转金属和还原消除三个步骤共同影响,其微妙的相互作用可能是Suzuki偶联较类似的Negishi偶联对修饰底物耐受性低的原因。
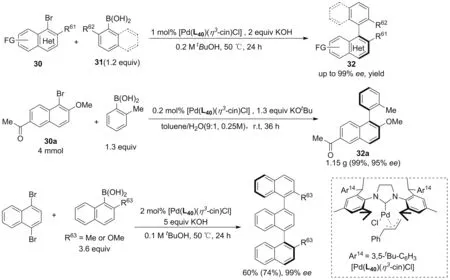
2019年,Shi课题组[59]开发了大位阻富电子的NHC卡宾钯催化剂[Pd(L40)(η3-cin)Cl],在高效高对映选择性合成官能团化双芳基、异二芳,三联萘等化合物上取得了阶段性成果(Scheme 23)。该催化体系的底物范围非常广泛,具有反应活性和对映选择性优异、催化剂负载低(1 mol%)、条件温和等优势。

Scheme 24
Scheme 25
5.2 钯催化的非对映选择Suzuki偶联
2003年,Colobert课题组[60]首次开发了β-甲氧基对甲苯亚砜手性辅助的芳基卤化物33与邻取代芳基硼酸(酯)的不对称Suzuki偶联,de值高达98%(a,Scheme 24)。研究表明,硫手性基团在立体控制中必不可少,而手性碳中心对立体控制的贡献更大。之后,手性底物34也被用于与邻取代芳基硼酸(酯)的不对称Suzuki偶联[61](b,Scheme 24)。
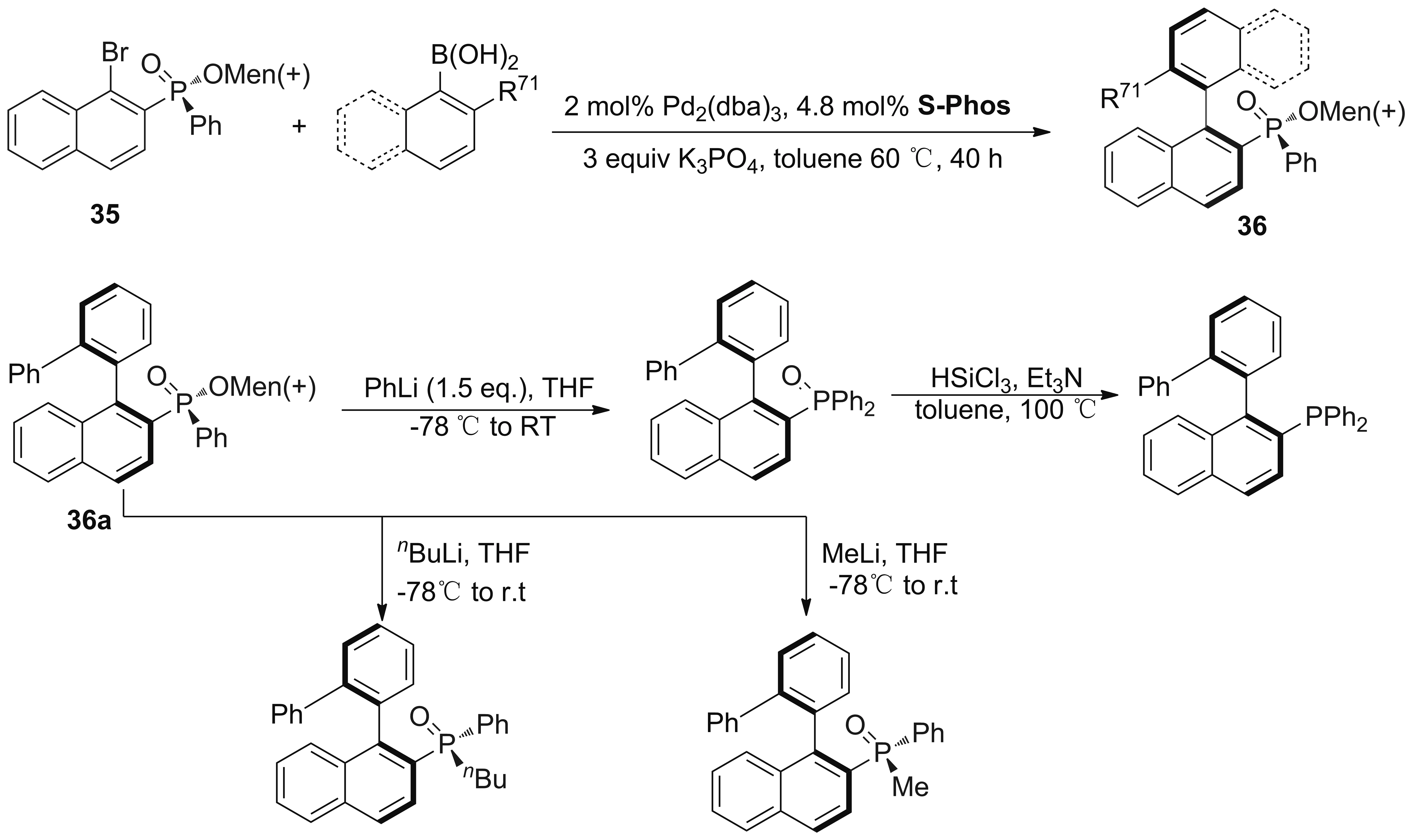
2015年,Yang课题组[62]尝试潜手性底物35与邻取代芳基硼酸的不对称Suzuki偶联,偶联产物36a在克级反应中能得到中等收率和90%的de值,可以进一步衍生成其他含膦化合物(Scheme 25)。
综述了镍、铜、钒、铁,钯等过渡金属催化的不对称Negishi偶联、氧化偶联、Suzuki偶联等经典直接偶联方法构建轴手性芳基-芳基化合物。不对称氧化偶联在各种联芳酚类化合物的构建中得到广泛应用;而钯催化的不对称Suzuki偶联反应底物适用性广、稳定性高、催化活性高,适用于各种轴手性、面手性、中心手性膦配体,其中构象良好的苯并五元氧膦膦中心手性配体显示出优异的反应活性和对映选择性,有些已成功用于天然产物和药物开发。除了膦配体外,手性NHC卡宾等也可作为钯催化剂的配体,其具有更大空间易于优化钯活性物种的手性环境,在不对称Suzuki偶联中有较大前景。然而,目前所用催化剂相对昂贵,廉价金属催化的不对称偶联构建轴手性芳基-芳基化合物值得期待。钯催化的不对称Suzuki偶联已合成一系列轴手性联芳基化合物,但2,2′-位具有小官能团的手性联芳化合物仍存在挑战。因此,寻找更高效、稳定且经济环保的配体或其他催化体系催化这类反应具有深远意义,同时极具挑战性。另外,近年来兴起的惰性化学键的活化反应(碳碳键的活化、碳氢键的活化、碳氮键的活化)、串联反应等策略在轴手性合成中的应用也将发挥更为重要的作用。
