集胞藻PCC6803中α-酮戊二酸脱羧酶的原核表达分析
2020-09-07刘志文王小琴李至敏谢琮琮李志敏
张 伟,刘志文*,王小琴,李至敏,谢琮琮,李志敏*
(1.江西农业大学 生物科学与工程学院,江西 南昌 330045;2.江西农业大学 理学院,江西 南昌 330045)
【研究意义】蛋白质是一切生命活动的物质基础,如何获得高纯度且保持活性的目的蛋白是深入研究生命活动的关键问题[1]。由于微生物操作简单、繁殖速度快等巨大优势,采用微生物生产活性蛋白的方法已被广泛应用于生物学和生物医学研究领域[2]。其中采用微生物进行异源表达重组蛋白是常用的手段,目前表达重组蛋白的系统可以分为细菌、酵母、丝状真菌、单细胞藻类、杆状病毒、昆虫细胞、植物细胞、哺乳动物细胞等,具体选择何种细胞来进行异源表达取决于目的蛋白[3]。【前人研究进展】大肠杆菌表达系统是目前发展最为完善也是最常用的高效异源表达重组蛋白的原核表达系统[4]。Page 等[5]研究了在大肠杆菌中以最经济的方式实现最大化表达异源蛋白的策略。Ceccarelli 等[6]综述了大肠杆菌异源表达重组蛋白的诸多方案和面临的挑战。然而,大肠杆菌表达系统也存在诸多缺点。例如,利用大肠杆菌异源表达真核生物来源的蛋白时不能进行翻译后修饰,不能充分形成二硫键导致无法获得可溶性蛋白等。因此很多在大肠杆菌表达系统中成功异源表达的蛋白以包涵体的形式存在。为了提高重组蛋白在大肠杆菌表达系统中的可溶性,研究人员开发了多种助溶标签,如硫氧还蛋白(thioredoxin,Trx)、谷胱甘肽巯基转移酶(glutathione S-transferase,GST)、麦芽糖结合蛋白(maltose binding protein,MBP)及小分子泛素样修饰蛋白(small ubiquitin-like modifier,SUMO)等[7-10]。其中一些标签还有助于重组蛋白的分离纯化。除此之外,研究发现DnaK、DnaJ、GrpE、GroES、GroEL、tig 等分子伴侣蛋白可以帮助新生蛋白质组装和正确折叠,从而提高重组蛋白可溶性,甚至提高其稳定性和活性[11-13]。【本研究切入点】α-酮戊二酸脱羧酶(α-KGD,EC 4.1.1.71)催化α-酮戊二酸转化为琥珀酸半醛,琥珀酸半醛在琥珀酸半醛脱氢酶的催化作用下转化为琥珀酸从而进入三羧酸循环。由于蓝细菌中缺乏α-酮戊二酸脱氢酶,因此α-KGD 就成了蓝细菌三羧酸循环中关键酶之一[14]。目前关于α-KGD的研究非常少,最新研究也仅停留在催化动力学上,其结构功能关系和催化机制研究均未有报道[15]。本课题组发现多种蓝细菌来源的α-KGD 在大肠杆菌表达系统中进行异源表达后在裂解液上清中的可溶性不高,主要以包涵体的形式存在于沉淀中,非常难以获得大量的可溶性α-KGD,这可能是造成目前未有研究报道有关α-KGD 的结构功能关系及催化机制的重要原因之一。【拟解决的关键问题】在NCBI 数据库中集胞藻PCC6803 中的sll1981基因被标注为编码乙酰乳酸合成酶。然而本课题组通过氨基酸序列比对发现sll1981基因编码蛋白和聚球藻PCC7002 中的a2770基因编码蛋白的氨基酸序列一致性达到80%。聚球藻PCC7002 中的a2770 蛋白已被证实为一个α-KGD[14]。为了阐明集胞藻PCC6803 中的sll1981基因编码蛋白的功能,首先就要获得可溶性的重组sll1981 蛋白。因此本试验以来自集胞藻PCC6803 中的sll1981基因编码蛋白为研究对象,构建了含有sll1981基因的多种表达载体,探索了这些表达载体在大肠杆菌表达系统中的表达和可溶性情况,并比较不同方法表达后分离纯化得到的sll1981 蛋白催化活性。动力学测试表明集胞藻PCC6803 中sll1981基因编码蛋白为α-KGD。本研究为在大肠杆菌中异源表达α-KGD 提供了重要信息,也将有助于该酶的结构功能关系及催化机制的进一步研究。
1 材料和方法
1.1 材料
1.1.1 质粒和菌株 集胞藻PCC6803 基因组DNA、pET-28b 质粒、pET-32a 质粒、pCold-TF 质粒和分子伴侣pTf16质粒由本实验室保藏;大肠杆菌BL21(DE3)感受态细胞购自北京全式金生物技术有限公司。
1.1.2 主要试剂 T4 DNA 连接酶、DNA Marker 购自北京全式金生物技术有限公司;2×PfuPCR Master-Mix、质粒小提试剂盒和琼脂糖凝胶回收试剂盒购自天根生化科技(北京)有限公司;PageRuler 预染蛋白Ladder 购于ThermoFisher Scientific 公司;限制性内切酶BamHI、XhoI 和NdeI 购自NEW ENGLAND BioLabs;牛凝血酶(Thrombin)购于Sigma-Aldrich公司;Ni-NTA Agarose 购自QIAGEN公司;其他化学试剂均为分析纯,购于北京索莱宝生物科技有限公司;鱼腥藻PCC7120来源的琥珀酸半醛脱氢酶(ApSSADH)由本实验室纯化。
1.2 PCR扩增sll1981基因
以集胞藻PCC6803 基因组DNA 为模板扩增sll1981基因,使用ApE 软件设计上游引物sll1981-F(AAATTTGGATCCATGGGGCAAATGAACACCGCAG)和下游引物sll1981-R(AATAATCTCGAGTTATT CCCAAATTTCACAGGCC)(序列中下划线为相应酶切位点)。反应体系包含基因组1 µL(终浓度为30 ng/µL),sll1981-F 和sll1981-R 各1µL(终浓度为0.2µmol/L),2×PfuPCR MasterMix 为25µL,加超纯水22µL 至总体积50µL。扩增反应条件为94 ℃预变性5 min,94 ℃变性30 s,55 ℃退火30 s,72 ℃延伸2 min,30 个循环,最后72 ℃延伸10 min[16]。PCR 反应结束后得到的产物经1.5%(m/v)琼脂糖凝胶电泳,然后切胶纯化PCR产物。
1.3 含有sll1981基因的表达质粒的构建
回收得到的30µL PCR 产物中加入10×buffer 5µL,NdeI 和XhoI 限制性内切酶各1µL,置于37 ℃水浴恒温酶切1 h。同理,用相应的限制性内切酶对pET-28b载体进行消化。胶回收酶切片段后用T4 DNA连接酶于16 ℃恒温水浴下过夜连接(目的片段与载体片段的摩尔比为9∶1)。次日取连接液10µL 转化大肠杆菌BL21(DE3)感受态细胞,然后再涂布于卡那霉素抗性平板上筛选,次日挑取单克隆菌落做PCR鉴定和DNA 测序鉴定。将测序正确的质粒保存为pET28b-sll1981质粒。用同样的方法构建pET32asll1981质粒和pColdTF-sll1981质粒。
1.4 重组sll1981蛋白诱导表达条件的优化
1.4.1 重组sll1981蛋白的诱导表达 挑取转化有pET28b-sll1981质粒的单克隆菌落于含有50µg/mL卡那霉素的LB培养液中过夜培养,次日按体积分数1%(v/v)接种量加入新鲜LB培养液中进行菌体扩大培养。待培养液OD600至0.6~0.8 时立即冰水浴30 min,然后加入终浓度为0.2 mmol/L 异丙基硫代半乳糖苷(IPTG)诱导剂于16 ℃、180 r/min条件下诱导表达24 h。次日以6 000 r/min的转速离心收集菌体,得到的菌体用20 mmol/L pH 7.5 的Tris-HCl 缓冲液重悬,随后置于冰水浴中超声破碎(变幅杆Φ 为10 mm,超声工作2 s,间歇8 s,有效功率10%(W/W),工作总时间5 min)。细胞破碎液于4 ℃、12 000 r/min离心30 min得到上清液和沉淀,最后电泳检测sll1981蛋白表达情况。
1.4.2 诱导温度的优化 采用上述方法培养转化有pET28b-sll1981质粒的大肠杆菌菌体,在加入0.2 mmol/L IPTG 后,将菌体分别置于16 ℃、20 ℃和25 ℃条件下诱导表达24 h,最后电泳检测sll1981 蛋白表达情况。
1.4.3 诱导时间的优化 采用上述同样的方法培养转化有pET28b-sll1981质粒的大肠杆菌菌体,加入0.2 mmol/L IPTG 后将菌体置于16 ℃、180 r/min 的恒温摇床培养箱中分别诱导表达6,12,24 h,最后电泳检测sll1981蛋白表达情况。
1.4.4 诱导剂浓度的优化 采用上述同样的方法培养转化有pET28b-sll1981质粒的大肠杆菌菌体,在菌体OD600至0.6~0.8 时分别加入终浓度为0、0.02、0.05、0.1、0.2、0.5 和1.0 mmol/L 的IPTG,然后将菌体均置于16 ℃、180 r/min条件下诱导表达24 h,最后电泳检测sll1981蛋白表达情况。
1.4.5 溶氧量的优化 将转化有pET28b-sll1981质粒的菌体过夜培养液分别接种于含有100,200,300,400 mL新鲜LB培养液的1 L锥形瓶中进行菌体扩大培养,待培养液OD600至0.6~0.8时,立即冰水浴30 min,再向其中加入0.2 mmol/L IPTG,置于16 ℃、180 r/min 条件下诱导表达24 h,最后电泳检测sll1981 蛋白表达情况。
1.5 重组sll1981蛋白的自诱导表达
将转化有pET28b-sll1981表达质粒和pET-28b 对照质粒的大肠杆菌BL21(DE3)细胞分别于含有50µg/mL 卡那霉素的LB 培养液中恒温过夜培养。次日,用4 个灭菌处理后的离心管分别离心收集体积分数1%接种量的菌体,接着用自诱导培养液重悬收集的细胞,再将细胞接种至100 mL 含有100µg/mL卡那霉素的新鲜自诱导培养液中,自诱导培养液按照文献配置[17]。然后将上述菌体置于16 ℃、180 r/min条件下分别自诱导表达16,20和24 h。最后电泳检测自诱导表达sll1981蛋白情况。后文将用此法获得的sll1981蛋白标注为Auto-sll1981。
1.6 重组sll1981蛋白与分子伴侣pTf16质粒共表达
采用CaCl2法制得转化有分子伴侣质粒pTf16 的大肠杆菌BL21(DE3)感受态细胞,再将pET28bsll1981质粒转化至该感受态细胞。挑取菌落加入到同时含有50µg/mL 卡那霉素和25µg/mL 氯霉素的LB 培养液中过夜培养。次日按体积分数1%接种量接种至含终浓度为1 mg/mL L-(+)-阿拉伯糖的新鲜LB 液体培养基(含上述相应浓度抗生素)中进行菌体扩大培养。待培养液OD600约为0.8 时冰水浴冷却30 min,然后加入终浓度为0.2 mmol/L IPTG,置于16 ℃、180 r/min 条件下诱导表达24 h。最后电泳检测分子伴侣tig蛋白与sll1981蛋白共表达情况。后文将用此法获得的sll1981蛋白标注为Co-sll1981。
1.7 不同表达质粒上sll1981蛋白的诱导表达
将上述构建成功的pET28b-sll1981、pET32a-sll1981和pColdTF-sll19813 种质粒分别转化大肠杆菌BL21(DE3)感受态细胞。挑取单菌落分别于含50µg/mL 卡那霉素、100µg/mL 氨苄青霉素和100µg/mL氨苄青霉素的LB培养液中过夜培养。次日按体积分数1%接种量分别加入100 mL新鲜LB培养液(含上述相应浓度抗生素)中进行菌体扩大培养,待培养液OD600至0.6~0.8时立即冰水浴30 min,然后向其中加入终浓度为0.2 mmol/L IPTG,于16 ℃、180 r/min 条件下诱导表达24 h。最后电泳检测sll1981 蛋白表达情况。后文分别标注为sll1981、Trx-sll1981和TF-sll1981。
1.8 重组sll1981蛋白的分离纯化
利用镍亲和层析的方法对上述sll1981 蛋白、Auto-sll1981 蛋白、Co-sll1981 蛋白、Trx-sll1981 蛋白和TF-sll1981 蛋白进行分离纯化。以纯化Auto-sll1981 蛋白为例。超声波破碎细胞后于4 ℃、12 000 r/min条件下离心1 h,得到的上清液用0.45µm滤膜过滤后流穿经20 mmol/L Tris-HCl,pH 7.5缓冲液平衡后的Ni-NTA柱,然后用含20~200 mmol/L咪唑的20 mM Tris-HCl,pH 7.5缓冲液进行梯度洗脱,分别收集不同浓度下的流穿液并电泳检测。合并高浓度高纯度sll1981 蛋白的洗脱液后于4 ℃浓缩(PEG 20 000)脱盐(透析3 次)处理,得到的sll1981 蛋白直接用于酶活测定。采用相同的方法纯化另外3 种sll1981 蛋白。另外,取纯化得到的部分TF-sll1981 蛋白于4 ℃经牛凝血酶酶切4 h,然后重新流穿平衡后的Ni-NTA 柱以除去标签,最后得到无TF标签的sll1981蛋白。通过测量蛋白质在280 nm处的吸光度,计算出无TF标签的sll1981蛋白(ε=58 955 L/(mol·cm))和TF-sll1981蛋白的浓度(ε=35 995 L/(mol·cm))。
1.9 重组sll1981蛋白的活性检测
以α-酮戊二酸(α-KG)为底物,采用酶偶联法分别检测自诱导表达后纯化得到的sll1981蛋白、与分子伴侣pTf16质粒共表达后纯化得到的sll1981蛋白、带TF标签的sll1981蛋白以及无TF标签的sll1981蛋白的活性[18]。500µL标准反应体系中包含100 mmol/L Tris-HCl,pH 7.5缓冲液,2 mmol/L Mg2+,2 mmol/L焦磷酸硫胺素(TPP),2 mmol/L 烟酰胺腺嘌呤二核苷酸磷酸(NADP+),α-KG 浓度为2~30 mmol/L,9µmol/L 来源于鱼腥藻PCC7120 的琥珀酸半醛脱氢酶(ApSSADH)[19],5µmol/L sll1981 蛋白。使用紫外分光光度计连续测定反应混合液在340 nm 处的吸光值(A340)变化,并根据NADPH 的生成速率计算出加入不同浓度α-KG 时的初始速度。运用KaleidaGraph 4.0 软件绘制动力学曲线,并根据米氏方程v=vmax·[S]/(Km+[S])计算出Km和kcat。试验重复3次。
2 结果与分析
2.1 重组pET28b-sll1981质粒的构建与鉴定
PCR 扩增产物经琼脂糖凝胶电泳后获得一条位于1.5~2.0 kbp,大小约1.7 kbp 的DNA 条带(图1A)。该基因片段与sll1981基因理论大小(1 653 bp)相吻合。将该扩增得到的sll1981基因酶切后和用相应酶切后的pET-28b 载体按比例连接并转化大肠杆菌BL21(DE3)感受态细胞,次日随机挑取7 个单克隆菌落进行PCR鉴定。如图1B可知,3、4、5、7和8号条带为阳性克隆,2和6号条带为假阳性克隆。选取其中3 个阳性克隆点送由上海祥音生物科技有限公司进行DNA 测序,测序结果经ApE 软件分析证明PCR 扩增获得到的sll1981基因序列与NCBI 数据库中的sll1981基因序列完全一致。因此,成功构建得到pET28b-sll1981表达菌株。
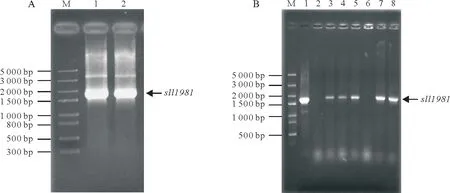
图1 集胞藻PCC6803的sll1981基因PCR扩增和菌落PCR鉴定Fig.1 Amplification and clone identification of sll1981 gene from Synechocystis sp.PCC6803
2.2 重组sll1981蛋白的表达条件的优化
重组pET28b-sll1981质粒转化大肠杆菌后在0.2 mmol/L IPTG,16 ℃、180 r/min 条件下诱导表达24 h后的结果如图2所示。和空载体表达情况比较发现,pET28b-sll1981表达菌株在该条件下成功诱导表达大量sll1981 蛋白,然而诱导表达的sll1981 蛋白在裂解缓冲液中基本不可溶。随后,本研究探索了各项诱导表达条件,以期找到最佳诱导表达条件,方便后续蛋白纯化以及活性测试。
首先探索了诱导温度的影响,结果如图3 所示。重组sll1981 蛋白在16 ℃、20 ℃和25 ℃下表达量均很高,然而在裂解缓冲液中的可溶性很差,低温并没有改善其可溶性。同时,研究发现当诱导表达温度为20 ℃和25 ℃时,重组sll1981蛋白的表达量比16 ℃时稍高。
其次探索了诱导时间的影响,其表达结果如图4A所示。由图4A可知诱导相对较长时间(24 h)可以显著提高sll1981 蛋白的表达量,诱导时间过短(6 h)会使得sll1981 蛋白表达量大大减少,然而诱导时间并不影响sll1981 蛋白的可溶性。本研究还探索了IPTG 浓度的影响。如图4B 所示,当不添加IPTG 时,sll1981 蛋白没有表达。当添加IPTG 浓度在0.02~1 mmol/L 时,sll1981 蛋白的表达量没有明显差别。在此条件表达时,重组sll1981蛋白没有明显的可溶性。此外本研究还探索了培养液的溶氧量对sll1981蛋白表达和可溶性的影响。其诱导表达情况如图4C。研究发现,溶氧量的改变并没有显著影响sll1981蛋白的表达量和可溶性。
2.3 其他含有sll1981基因重组质粒的表达
在大肠杆菌中自诱导表达外源基因获得重组蛋白的方法简便易操作,因此本研究也探索了自诱导表达方式对pET28b-sll1981重组质粒在大肠杆菌中表达重组sll1981 蛋白的影响。结果表明,在16 ℃时自诱导16 h没有诱导表达sll1981蛋白,自诱导20 h后该菌株表达了非常少量的sll1981蛋白。当诱导时间达24 h 后sll1981 蛋白被大量表达。因此,采用自诱导培养基也能有效表达重组sll1981 蛋白,然而此方法表达的sll1981 蛋白可溶性并没有显著提高(图5A)。此前研究表明过表达分子伴侣蛋白可以促进目的蛋白的折叠,从而提高重组蛋白的可溶性。本研究采用pET28b-sll1981重组质粒与分子伴侣质粒pTf16共转化大肠杆菌表达sll1981蛋白的方式,其结果如图5B所示。分子伴侣质粒pTf16表达的触发因子tig 蛋白大小为56 ku,比目的蛋白sll1981 小,与电泳结果中显示的tig 蛋白处于sll1981 蛋白下方一致,下图接近55 ku的蛋白分子即为触发因子tig蛋白。由图5B可知,与分子伴侣质粒pTf16共表达可以显著提高sll1981 蛋白的可溶性。同时笔者也构建了pET32a-sll1981和pColdTF-sll1981重组质粒,其分别含有可能提高重组蛋白可溶性的Trx 和触发因子TF 标签。研究结果表明sll1981 蛋白连接Trx 标签后其可溶性没有显著提高,然而sll1981 蛋白连接TF 标签后其可溶性显著提高,表达的sll1981 蛋白基本都在上清中(图5C)。
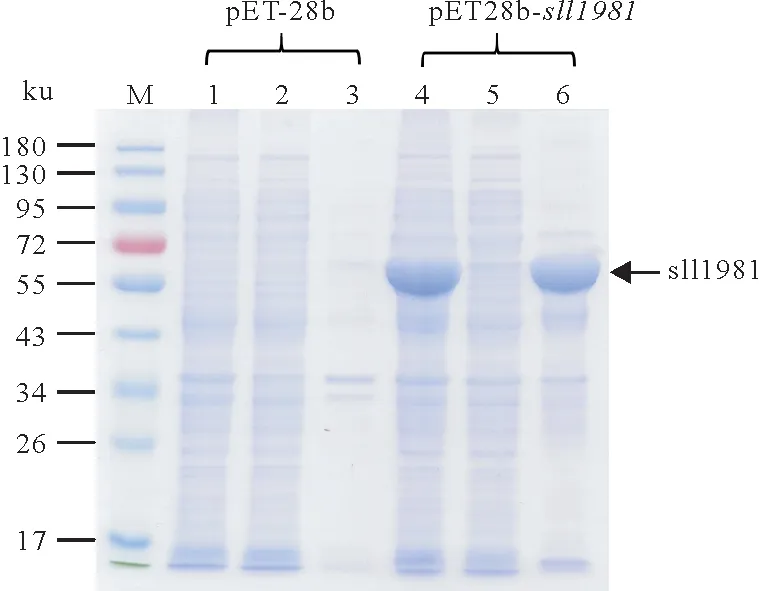
图2 重组pET28b-sll1981质粒在大肠杆菌中的诱导表达Fig.2 The induced expression of recombinant sll1981 protein in Escherichia coli
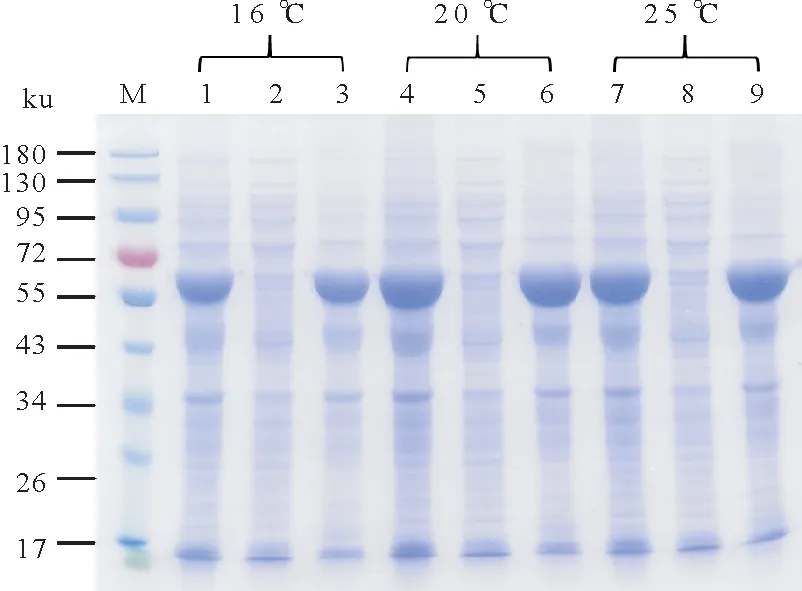
图3 不同温度下重组sll1981蛋白诱导表达Fig.3 Induced expression of recombinant sll1981 protein at deferent temperatures
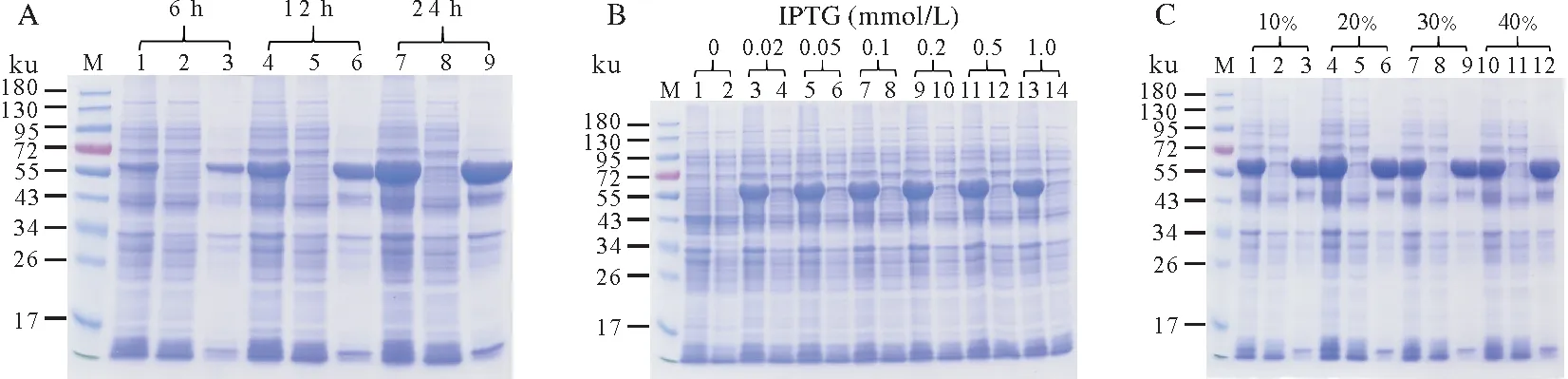
图4 重组pET28b-sll1981质粒在不同条件下的诱导表达Fig.4 The expression of recombinant pET28b-sll1981 plasmid at various conditions
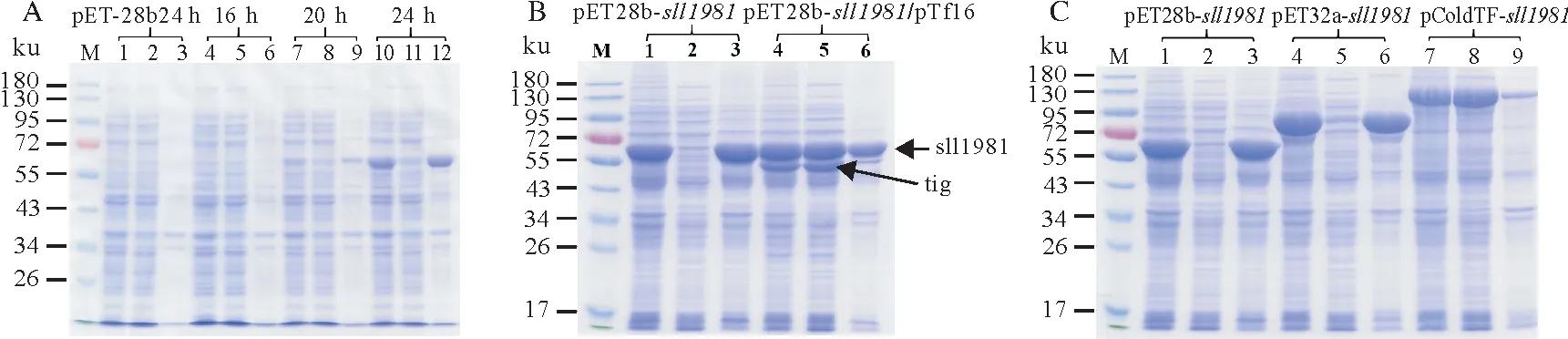
图5 重组sll1981蛋白的表达情况Fig.5 The expression of recombinant sll1981 protein at various vectors
2.4 重组sll1981蛋白的纯化
分别对上述自诱导(Auto-sll1981)、与pTf16 共表达(Co-sll1981)和TF-sll1981 融合表达(TFsll1981)方法表达的重组sll1981蛋白进行分离纯化。此前研究发现当纯化IPTG诱导表达的sll1981蛋白时,洗出的重组sll1981容易形成白色浑浊沉淀,因此最终未能纯化得到sll1981蛋白。随后尝试对Autosll1981 蛋白进行分离纯化(图6A),最终得到浓度为10µmol/L 的sll1981 蛋白(图6B)。纯化过程未见明显白色浑浊,可推测自诱导表达的sll1981 蛋白相对较稳定。对Auto-sll1981 浓缩时发现当Auto-sll1981浓度超过10µmol/L时该蛋白还是会聚集形成白色沉淀。对Co-sll1981蛋白进行分离纯化(图7A),最终得到浓度为30µmol/L的sll1981蛋白(图7B)。采用与pTf16质粒共表达后纯化得到的Co-sll1981蛋白浓度明显高于自诱导表达后纯化得到的Auto-sll1981蛋白浓度,由此推测Co-sll1981蛋白较上述两种方法表达的sll1981蛋白更加稳定。

图6 自诱导表达sll1981蛋白的纯化Fig.6 Purification of auto-induced expression of recombinant sll1981 protein
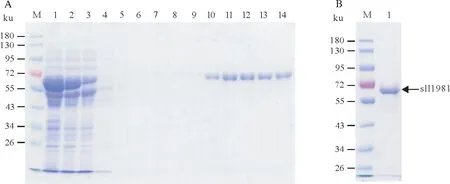
图7 与pTf16分子伴侣共表达sll1981蛋白的纯化Fig.7 Purification of sll1981 protein co-expressed with chaperone plasmid pTf16
重组TF-sll1981 蛋白的纯化如图8 所示。由于得到的TF-sll1981 蛋白带有分子量较大的TF 标签,因此将TF-sll1981 蛋白于4 ℃经凝血酶酶切后再次流穿Ni-NTA 亲和层析柱后使得sll1981 蛋白与TF 标签蛋白分离,得到无TF标签的高纯度sll1981蛋白(图8B)。
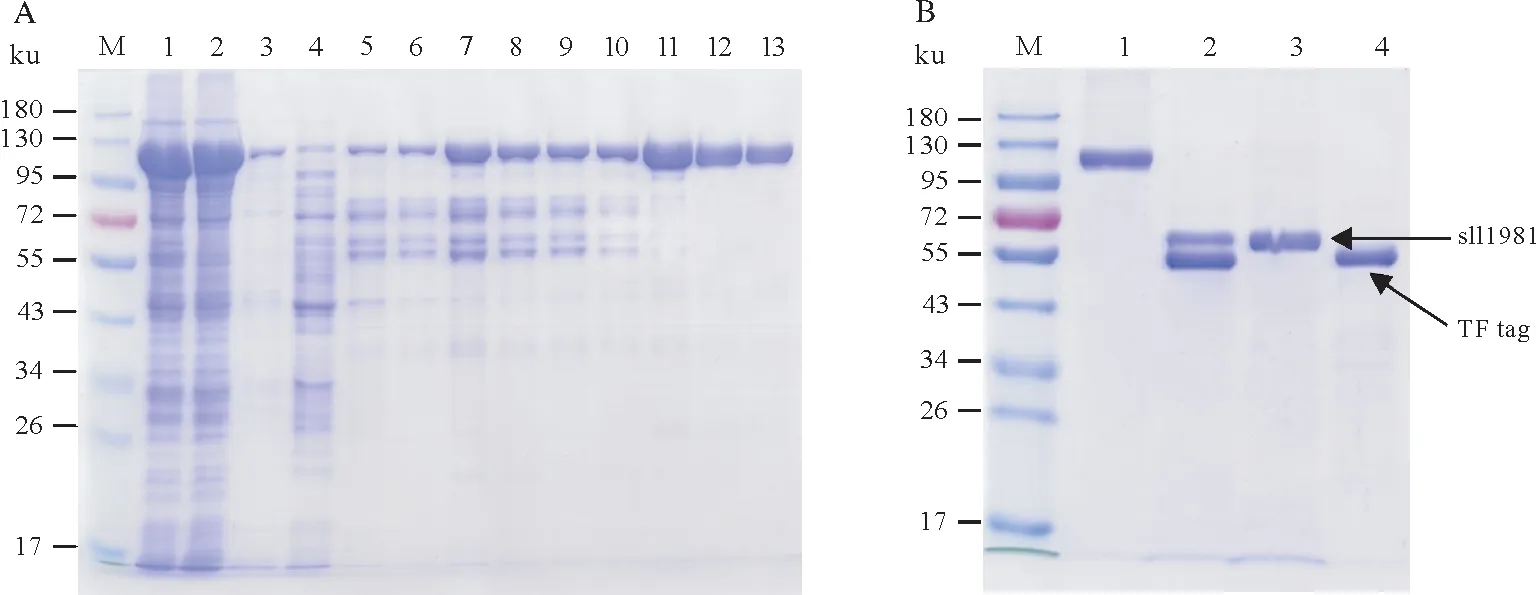
图8 与TF标签融合表达sll1981蛋白的纯化结果Fig.8 Purification of sll1981 protein expressed with TF tag
2.5 重组sll1981蛋白作为α-KGD的活性比较
由于sll1981蛋白非常不稳定,经IPTG诱导表达的sll1981蛋白在用咪唑梯度洗脱时已经形成白色沉淀,因此无法检测其催化活性。其他方法纯化得到的sll1981蛋白均能检测出α-KGD活性(表1)。研究结果表明催化活性最高的sll1981蛋白是采用与pTf16质粒共表达的Co-sll1981蛋白,其次为Auto-sll1981蛋白,活性最低的是TF-sll1981蛋白。当TF-sll1981蛋白的TF标签被酶切后,尽管笔者可以纯化得到不带TF标签的sll1981蛋白,然而在测定活性的缓冲液体系中该不带TF标签的sll1981蛋白极易沉淀,因此无法测定其催化活性。当底物α-KG浓度超过6 mmol/L时,采用自诱导方式获得的Auto-sll1981在活性测定缓冲体系中也易沉淀,因此本试验仅能测定其在底物浓度为6 mmol/L时的反应速度为0.670µmol/(L·s),无法测得其他动力学参数。
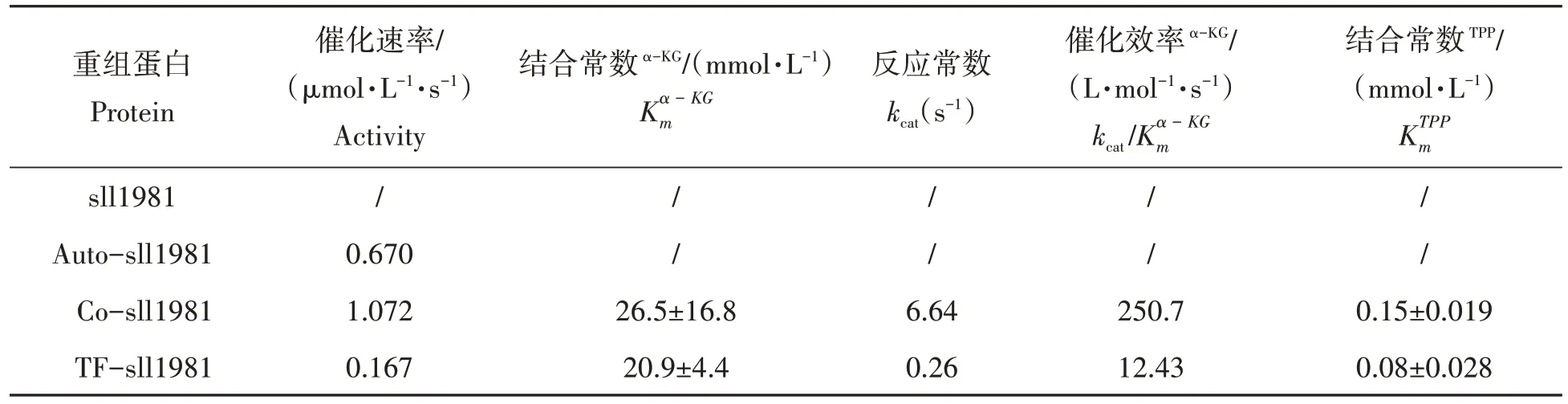
表1 不同方法表达的重组sll1981蛋白活性比较Tab.1 Activity comparison of recombinant sll1981 protein expressed by different methods
3 讨论与结论
此前研究表明菌株、培养基、诱导温度、IPTG 浓度及诱导时间均会影响重组蛋白在异源表达系统中的表达量和可溶性。例如,Biria 研究组[20]通过taguchi 法优化诱导温度和时间及培养基配方大大提高了人类生长激素蛋白(hGH)在大肠杆菌Origami 菌株中的产率。Xu 等[21]通过优化IPTG 浓度和诱导时间,使得重组疏水蛋白(HGFI)在大肠杆菌BL21 菌株中得到了满意的表达。本研究比较了pET28b-sll1981质粒在大肠杆菌表达系统中不同温度,IPTG 浓度及诱导时间、不同溶氧体积比及自诱导培养基中的表达情况。研究发现重组sll1981 蛋白在大肠杆菌BL21(DE3)菌株中表达量很高,然而基本都是以包涵体形式存在。通过与Trx 助溶标签融合表达的方式也无助于提高sll1981 蛋白的可溶性。尽管与pColdTF 质粒中TF 标签融合表达后提高了sll1981 蛋白的可溶性,但是酶切TF 标签蛋白后sll1981 蛋白非常不稳定,因此与TF 标签融合表达这种方法也不可行。幸运的是通过与pTf16 分子伴侣质粒共表达的方式获得了可溶性的sll1981 蛋白,并且具有较高的催化α-KG 脱羧生成琥珀酸半醛的活性。本研究数据为生产可溶性具有催化活性的sll1981 蛋白质提供了重要基础。事实上,2006 年Chatterjee 等人发现pET21b-sll1981质粒转化大肠杆菌BL21 菌株后表达的sll1981 蛋白也是以包涵体形式存在,该作者通过包涵体复性的方法纯化得到sll1981 蛋白,但是用此法获得的sll1981 没有α-KGD 活性[22]。本实验室未发表的数据发现与sll1981 蛋白氨基酸序列高度一致的其他蓝细菌来源的α-KGD 在大肠杆菌表达系统中的可溶性均很差,因此本研究结果对于纯化其他来源的α-KGD 也有重要的参考意义。
此前研究指出自诱导培养基较普通LB 培养基具有均匀的高细胞培养密度和较强的表达能力[23]。因此,本研究尝试了自诱导培养基表达重组sll1981 蛋白。SDS-PAGE 检测发现表达的sll1981蛋白有少量存在于上清液中,继而对该方法表达的sll1981 蛋白进行纯化以及活性测试。在纯化过程中,洗出的sll1981 蛋白并没有形成沉淀,最终纯化得到浓度为10 µmol/L 的sll1981 蛋白,超过此浓度时蛋白极易发生沉淀。在测定该法获得的sll1981 蛋白活性时,最高仅能检测到底物α-KG 浓度为6 mmol/L,当超过该浓度时,sll1981 蛋白会聚集形成白色浑浊。猜想可能的原因有二。其一,当底物浓度增大时,反应液中离子强度发生改变促使sll1981 蛋白构象由稳定状态转向不稳定状态;其二,α-KG 含有两个羧基,当加入更高浓度的α-KG 时,反应液的pH 值有所改变,致使sll1981 蛋白逐渐形成白色沉淀。
分子伴侣蛋白有助于防止新生蛋白在细胞拥挤的环境中发生错误折叠和聚集[24]。其中触发因子TF(伴侣蛋白)会与核糖体1:1 结合,并与新生蛋白链相互作用,使其处于可进行后续折叠的状态[25-26]。因此本研究选用pTf16 分子伴侣质粒(表达的伴侣蛋白为TF)与pET28b-sll1981质粒在大肠杆菌中共表达,结果得到大量可溶性sll1981 蛋白,并且纯化得到较高浓度的sll1981 蛋白(30 µmol/L)。该结果说明在此浓度下sll1981 蛋白不会聚集形成沉淀,比自诱导表达的sll1981 蛋白更稳定。研究发现大肠杆菌硫氧还蛋白Trx 和触发因子TF 在大肠杆菌细胞质基质中可以防止初生蛋白错误折叠和聚集,使得多亚基蛋白不易形成包涵体,从而有效提高蛋白质的溶解性,因而开发了目的蛋白与标签蛋白融合表达的方法。本研究构建了两种融合表达质粒pET32a-sll1981和pColdTF-sll1981。结果显示sll1981蛋白与Trx 融合表达后溶解性并没有显著变化,而与TF 标签融合表达后的溶解性显著提高,并且纯化得到更高浓度的sll1981 蛋白(30 µmol/L)。遗憾的是TF-sll1981 蛋白酶切去TF 标签后获得的sll1981蛋白同样不稳定。
本研究探索了不同表达条件和表达方法对集胞藻PCC6803来源的sll1981蛋白表达量和可溶性的影响,最后纯化得到具有催化活性的重组sll1981 蛋白。酶动力学测试数据表明sll1981 蛋白具有α-KGD活性,并且不同表达方法获得的sll1981 蛋白的催化活性差异较大,其中当pET28b-sll1981质粒与pTf16分子伴侣质粒在大肠杆菌中共表达时,重组sll1981蛋白的可溶性和催化活性均为最佳。
